
缺血性脑卒中差异表达基因筛选与生物信息学分析
2023-12-14 02:23李晴晴李小霞张毓洪常晓玉
宁夏医科大学学报 2023年10期
李晴晴, 李小霞, 赵 燚, 张毓洪, 常晓玉,2
(1.宁夏医科大学公共卫生学院,银川 750004; 2.宁夏医科大学学报编辑部,银川 750004)
脑卒中是一种急性脑血管疾病,是由于脑部血管突然破裂或因血管阻塞导致血液不能流入大脑而引起脑组织损伤的一种疾病,包括缺血性脑卒中和出血性脑卒中,具有发病率高、致残率高、病死率高和复发率高的特点[1]。调查显示,中国脑卒中患病率随着年龄增长而升高且城市高于农村,2020 年总体患病率达到2.6%,成为我国第二位死亡原因,也是中国成年人残疾的首要原因[2]。目前公认的治疗脑卒中最有效的方法是溶栓治疗,但发病后4.5 h 内的治疗时间窗口狭窄且出血风险高,IS 患者的溶栓率非常低,预后差[3]。因此,确定有效治疗脑卒中的分子靶点和明确脑损伤的机制尤为重要。
研究[4]表明,IS 约占全部脑卒中的80%,心源性栓塞性卒中属于IS 的一种。IS 是一种多因素、多机制及多通路介导的疾病,其发病机制和关键基因尚不明确。有研究[5]表明,PTGS2 基因可能与小血管闭塞的IS 亚型有关;PITX2 和ZFHX3 基因以及另外2 个相关基因ZNF566 和PDZK1IP1的单核苷酸多态性会增加脑卒中的患病风险,不同表观遗传机制(如DNA 甲基化、组蛋白修饰、非编码RNA 等)可能诱导对脑缺血的不同损伤反应[6]。因此,对脑卒中风险位点的了解可增加找到新的抗血栓药物治疗靶点的可能性。而生物信息学分析可以利用高通量基因测序技术分析生物体的基因组、转录组和蛋白质组信息,从各个分子水平揭示疾病发生和发展的机制。本研究利用生物信息学方法选取IS 患者和正常人外周血标本全基因组表达的mRNA 芯片数据筛选IS 差异表达基因(differentially expressed genes,DEGs),分析其功能和信号通路,然后构建一个基因网络图,并筛选关键基因靶点,以期为临床诊断和治疗提供理论基础。
1 材料与方法
1.1 数据集获取
从美国国家生物技术信息中心(National Center for Biotechnology Information,NCBI)(https://www.ncbi.nlm.nih.gov/)基因表达综合数据库(gene expression omnibus,GEO),以“stroke”为检索词、“homo sapiens”为限定种属、“expression profiling by array”为限定类型,检索相关基因表达谱数据。纳入标准:1) 数据集是全基因组mRNA 芯片数据;2)数据样本来源于IS 患者和正常人外周血标本;3)独立数据集的标本量必须>20。
1.2 IS DEGs 筛选
利用GEO 数据库选定符合限定条件的芯片,下载基因原始数据的表达矩阵和检测平台信息。使用R 4.3.0 软件,利用DESeq2 包、edgeR 包和limma 包分别进行差异分析,DEGs 的筛选条件是P<0.05 且|log2FC|>0.5,其中差异倍数(fold change, FC)代表IS 患者与健康对照人群两者DEGs 的差值倍数。由于不同数据集的样本来源、采集时间等实验过程中的某些差异产生了影响研究结果的协变量(箱式图显示两个数据集的表达量不一致),故使用limma 包中的removeBatch-Effect 函数去除在基因表达数据中的批次效应。利用微生信(http://www.bioinformatics.com.cn/)在线分析平台进行Venn 分析和可视化分析。
1.3 GO 功能和KEGG 信号通路富集分析
利用R 语言clusterProfiler 包(4.8.1 版本)对DEGs 进行GO 功能和KEGG 信号通路富集分析,然后利用GO plot 包和ggplot 2 包进行富集结果的可视化。GO 功能富集分析包括生物过程(biological process,BP)、细胞组成(cellular component,CC)和分子功能(molecular function,MF)。
1.4 蛋白-蛋白相互作用网络
将DEGs 导入在线软件STRING 11.5(http://www.string-db.org/),对IS 发病相关基因表达产物构建PPI 网络。通过Cytoscape 3.9.1 软件分析结果制作可视化蛋白互作网络模型,并应用MCODE 插件筛选其中关联性强的蛋白互作模块,再利用cytoHubba 插件并依据平均度(degree)大小确定PPI 网络中的关键节点。
1.5 IS 关键基因的表达分析和诊断价值
取一个数据集进行LASSO 回归分析建模,所得基因与前面PPI 网络分析的Hub 基因进行交集得到关键的几个基因,最后利用ROC 曲线评估这些关键基因在IS 中的诊断价值。
1.6 统计学方法
将P<0.05 且FDR<0.05 作为GO 功能富集分析差异有统计学意义的信号通路;将P<0.05作为KEGG 信号通路富集分析差异有统计学意义的信号通路。LASSO 回归分析筛选IS 差异基因。通过ROC 曲线分析验证关键基因预测IS 的准确阈值。
2 结果
2.1 标本资料
根据纳入标准,选定符合条件的3 个GEO数据库(GSE22255[7]、GSE58294[8]和GSE16561[9])共195 个样本,采用GPL570 和GPL6883 平台检测。共纳入128 个IS 患者和67 个健康对照人群外周血标本,其中95 例男性、100 例女性,见表1。

表1 纳入标本的基本资料
2.2 DEGs 筛选
GSE22255 和GSE58294 数据集去除批次效应后合并(图1A、图1B),得到一个含有89 个IS组和43 个对照组的新数据集。再分别利用DESeq2 包、edgeR 包和limma 包,以P <0.05 且|log2FC|>0.5 为条件进行DEGs 的筛选(图1C、图1D、图1E)。DESeq2 包获得409 个上调基因和211 个下调基因,edgeR 包获得171 个上调基因和109 个下调基因,limma 包获得556 个上调基因和298 个下调基因,取三者的交集得到196 个DEGs(155 个上调基因和41 个下调基因),见图1F、图1G。

图1 差异基因的筛选
2.3 GO 功能和KEGG 信号通路富集分析
结果显示,共获得179 个差异显著的GO 条目,包括133 个BP 条目、41 个CC 条目和5 个MF 条目(P<0.05 且FDR<0.05)。GO 功能富集分析弦图显示,BP 中GO 条目最多的是应激反应,CC 中GO 条目最多的是内膜系统,MF 中GO 条目最多的是模式识别受体活化(图2A)。GO 功能分析太极图,显示DEGs 在总的GO 功能富集分析中基因富集最多的8 条GO 条目(4 条在BP中,4 条在CC 中)(图2B)。可知,DEGs 介导的生物过程主要集中在应激反应以及对刺激反应的调节等;在细胞组分方面,DEGs 主要分布在子宫内膜系统、囊泡等;在分子功能方面,DEGs 主要富集在模式识别受体活性、NAD+核苷酶活性等。KEGG 信号通路富集分析结果表明差异显著的途径有15 条,气泡图仅显示有显著意义的前10条路径(P<0.05)。由图2C 可知,DEGs 主要参沙门氏菌感染、NOD 样受体信号通路、造血细胞谱系等信号通路;并且,由多个DEGs 调控同一条信号传导通路,来介导疾病的发生过程。

图2 DEGs 的GO 功能和KEGG 途径富集分析
2.4 PPI 网络的构建和枢纽基因的选择
结果显示,共180 个节点的PPI 拓扑图,含168 条边,degree 为1.87,见图3A。使用Cytoscape软件绘制蛋白互作网络模型(含94 个节点,153条边),见图3B。选用Cytoscape 的MCODE 插件筛选PPI 网络中的核心模块,所得功能模块最大的一个子网络由6 个节点、12 条边组成,score 为4.8,该模块即PPI 网络中紧密联系的区域,见图3C。基于cytoHubba 插件,并根据degree 大小确定PPI 网络中的关键节点,得到排名前10 的枢纽基因(Hub 基因),见图3D。其中,排名前3 位的Hub 基因是基质金属蛋白酶9(matrix metallopeptidase 9,MMP9)、趋化因子受体7(C-C chemokine receptor 7,CCR7)、CD19 分子(CD19 molecule,CD19),MMP9 的degree(degree=18)高于其他蛋白节点。
2.5 IS 关键基因诊断价值的验证
GSE16561 数据集经DESeq2 包、edgeR 包和limma 包取交集所得的104 个DEGs 与前面所得的196 个DEGs 取交集得16 个DEGs,见图4A、4B。在GSE16561 数据集中,用R 语言的glmnet包对上述16 个DEGs 进行LASSO 回归分析,其中风险评分使用回归系数(coef)乘以基因表达值(exp)再进行相加,保留其中11 个基因时为最佳模型(图4C 和图4D)。这11 个差异基因对IS 总的ROC 诊断准确率为100%(图4E)。将这11 个差异基因与PPI 网络分析得到的10 个Hub 基因取交集最终得到4 个关键基因(图4F)。为验证这4 个关键基因预测IS 的准确率,构建了ROC曲线。CCR7、MMP9、Bcl2-相关蛋白A1(BCL2A1)和淋巴细胞抗原96(LY96)基因诊断IS 的准确率分别为85.0%、85.7%、70.5%、80.9%(图4G),进一步证实了这4 个基因具备预测IS 的特异度和灵敏度。并且在GSE16561 数据集中,CCR7 基因也属于下调基因,MMP9、BCL2A1 和LY96 基因的表达同样上调。
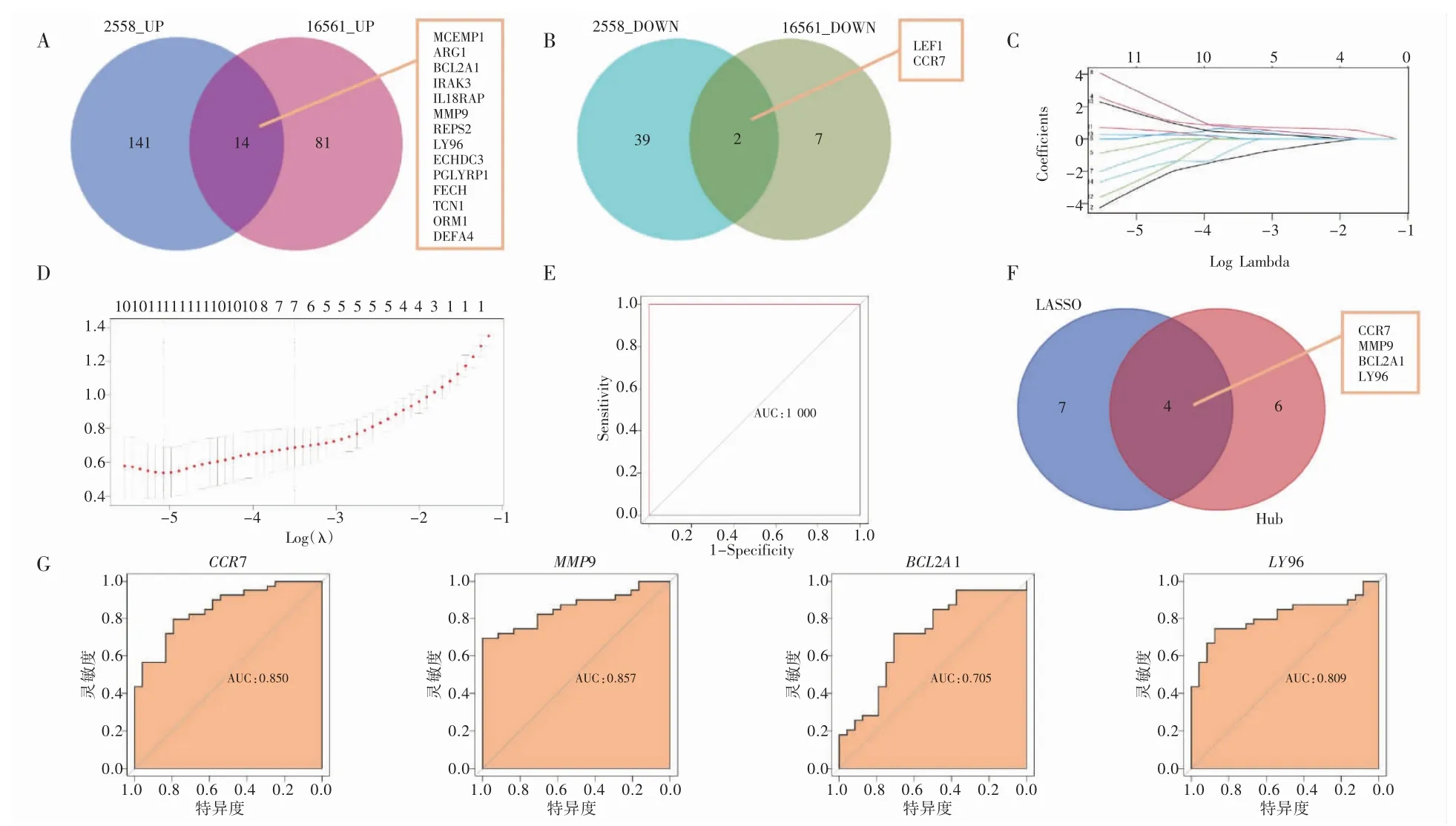
图4 IS 关键基因诊断价值的验证
3 讨论
本研究通过生物信息分析结果显示,CCR7、MMP9、BCL2A1 和LY96 基因在IS 患者的外周血中表达差异均有统计学意义,并且MMP9、BCL2A1和LY96 基因高度表达,可推测上述高表达的基因与IS 的发生率有关。同时,MMP9、BCL2A1 和LY96 基因编码的蛋白互作网络富集明显;共同参与涉及细胞表面受体、应激反应等信号通路。
MMP9 是目前研究最多的分泌型内肽酶之一,参与细胞外基质的局部蛋白水解、白细胞迁移和免疫反应等。MMP9 水平的升高与肿瘤患者的不良预后呈正相关,是乳腺癌、结直肠癌等肿瘤的潜在生物标志物[10-11]。 Wu 等[12]通过生物信息学和微阵列技术,鉴定MMP9 为急性心肌梗死的诊断标志物,且MMP9 与中性粒细胞、单核细胞、静息NK 细胞、γ-δ-T 细胞、CD4 记忆静息T细胞等免疫细胞均相关。 Li 等[13]利用定量逆转录PCR 和蛋白质印迹法证实了MMP9 是骨关节炎的潜在生物标志物。MMP9 也是IS 的重要调节因子。相关研究[14-16]表明,脑缺血时MMP9 表达升高,上调的MMP9 通过降解细胞外基质破坏脑微血管的结构完整性和血脑屏障,导致继发性脑水肿和脑损伤,而在敲除了小鼠的MMP9 或使用MMP9 抑制剂后,可减轻脑水肿。因此,MMP9有望成为治疗IS 的靶点。Yang 等[17]利用微阵列技术发现了在IS 患者中表达显著上调的多个生物标志物分子,MMP9 可能成为脑卒中患者诊断和预后的新分子靶点。后续仍需更多的实验研究来验证IS 中MMP9 的作用。
BCL2A1 是BCL2 家族的成员,凋亡过程由BCL2 家族蛋白控制,BCL2A1 可通过隔离促凋亡的BCL2 蛋白来发挥其抗凋亡功能[18]。BCL2A1 主要参与血管内皮细胞凋亡和动脉粥样硬化斑块的形成,其蛋白表达增加与银屑病表皮增生相关[19]。研究[20]发现,BCL2A1 可以同时预测结肠腺癌患者对化疗和免疫治疗的灵敏度,可能成为新的潜在化疗和免疫治疗靶点。但BCL2A1 作为IS 靶基因的相关报道较少,BCL2A1 在IS 中的作用机制尚不明确。本研究基于生物信息学理论发现,与对照组相比,BCL2A1 在IS 患者中高表达,其参与影响NF-κB 信号通路和癌症中的转录失调,可能在造血细胞对外部信号的反应和延缓白介素3(IL-3)剥夺诱导的细胞凋亡中起作用。而细胞凋亡在脑缺血中起主导作用,是脑缺血损伤后缺血半影区产生的主要病理生理机制之一[21],因此推测在IS 发生后,BCL2A1 可能通过影响造血系统和抑制细胞凋亡来减缓IS 的病情进展,建议BCL2A1 可作为IS 患者的潜在诊断和治疗靶点,其相关机制值得进一步探讨。
LY96 是免疫细胞识别脂多糖微生物结构成分的重要辅因子,与模式识别受体(pattern recognition receptors,PRRs)的关键成分Toll 样受体4(toll-like receptor4,TLR4)形成复合体,介导固有免疫应答[22]。LY96 的高表达已被证实与持续的促炎免疫反应相关,并可能参与修复肠道屏障功能破坏和调节菌群失调[23]。研究[9]表明,包含LY96 在内的一组免疫相关基因在IS 中高表达,并与外周血免疫系统功能高度相关[9]。脑卒中诱导的免疫抑制和随后的感染是患者死亡的主要原因[24]。脑卒中后免疫细胞凋亡的增加可能导致免疫抑制[25]。免疫细胞浸润可介导神经元凋亡,加重缺血性损伤[26]。因而,IS 的发生是多因素共同作用的结果,而细胞凋亡与免疫反应是其发生和发展中的重要环节。在本研究中,LY96 也在IS患者中高表达,主要参与脂多糖免疫受体活性、Toll 样受体结合等信号通路,其可能通过与TLR4 的协同作用介导对细菌脂多糖的先天免疫反应。因此推测IS 发生后诱导免疫抑制,随后LY96 通过促进先天免疫反应来减缓IS 的发生,建议LY96 也可以作为IS 患者的潜在诊断和治疗靶点,其相关机制值得进一步探讨。
综上所述,IS 患者的外周血发病过程中有多个基因表达发生了显著变化,最终筛选出来的MMP9、BCL2A1 和LY96 基因可能与IS 的发生有关联,可以作为今后对IS 干预的新研究靶点。但这些基因在IS 发病中的具体作用机制还未明确,以及本研究数据主要基于数据库生物信息分析得出,上述结论还需进行更深入的实验研究。
猜你喜欢
中老年保健(2021年3期)2021-12-03
中国生殖健康(2020年7期)2020-12-10
哈尔滨医药(2016年3期)2016-12-01
癌变·畸变·突变(2016年3期)2016-02-27
中国病理生理杂志(2015年8期)2015-12-21
医学研究杂志(2015年7期)2015-06-22
医学研究杂志(2015年3期)2015-06-10
创业家(2015年1期)2015-02-27
同位素(2014年2期)2014-04-16
遗传(2014年2期)2014-02-28
