
Dihydroartemisinin ameliorates innate inflammatory response induced by Streptococcus suis-derived muramidase-released protein via inactivation of TLR4-dependent NF-κB signaling
2023-12-14 08:31YunJiKaijiSunYingYangZhenlongWu
Journal of Pharmaceutical Analysis 2023年10期
Yun Ji ,Kaiji Sun ,Ying Yang ,Zhenlong Wu
a State Key Laboratory of Animal Nutrition and Feeding, China Agricultural University, Beijing,100193, China
b Beijing Advanced Innovation Center for Food Nutrition and Human Health, China Agricultural University, Beijing,100193, China
Keywords:Dihydroartemisinin Inflammation Muramidase-released protein Streptococcus suis Toll-like receptor 4
ABSTRACT Muramidase-released protein (MRP) is now being recognized as a critical indicator of the virulence and pathogenicity of Streptococcus suis (S.suis).However,the identification of viable therapeutics for S.suis infection was hindered by the absence of an explicit mechanism for MRP-actuated inflammation.Dihydroartemisinin (DhA) is an artemisinin derivative with potential anti-inflammatory activity.The modulatory effect of DhA on the inflammatory response mediated by the virulence factor MRP remains obscure.This research aimed to identify the signaling mechanism by which MRP triggers the innate immune response in mouse spleen and cultured macrophages.With the candidate mechanism in mind,we investigated DhA for its ability to dampen the pro-inflammatory response induced by MRP.The innate immune response in mice was drastically triggered by MRP,manifesting as splenic and systemic inflammation with splenomegaly,immune cell infiltration,and an elevation in pro-inflammatory cytokines.A crucial role for Toll-like receptor 4 (TLR4) in coordinating the MRP-mediated inflammatory response via nuclear factor-kappa B (NF-κB) activation was revealed by TLR4 blockade.In addition,NFκB-dependent transducer and activator of transcription 3(STAT3)and mitogen-activated protein kinases(MAPKs) activation was required for the inflammatory signal transduction engendered by MRP.Intriguingly,we observed an alleviation effect of DhA on the MRP-induced immune response,which referred to the suppression of TLR4-mediated actuation of NF-κB-STAT3/MAPK cascades.The inflammatory response elicited by MRP is relevant to TLR4-dependent NF-κB activation,followed by an increase in the activity of STAT3 or MAPKs.DhA mitigates the inflammation process induced by MRP via blocking the TLR4 cascade,highlighting the therapeutic potential of DhA in targeting S.suis infection diseases.
1.Introduction
The gram-positive bacteriumStreptococcus suis(S.suis) is most commonly associated with pigs,yet it is also capable of infecting humans and other mammals [1,2].S.suisbegins an infection by colonizing mucosal epithelial cells,after which it invades deeper tissues,and finally travels extracellularly to the bloodstream[3,4].A severe infection withS.suismay lead to an array of diseases including arthritis,sepsis,and meningitis.In addition,toxic shock syndrome,characterized by severe inflammation,is believed to be the most common precipitating factor for death linked toS.suis[4-6].
Despite several efforts,the mechanisms determiningS.suisvirulence remain elusive.Multiple virulence factors that are derived fromS.suishave been linked to the unfavorable outcomes of an infection caused byS.suis.Extracellular protein factor,muramidase-released protein (MRP),suilysin,and capsular polysaccharide (CPS) are all examples of such virulence factors [7-10].The cell wall-anchored fibrin binding protein MRP has been established as a virulence indicator forS.suisand is generally agreed upon as the singular virulence factor responsible for the pathogenicity ofS.suis[11].The D1 variable domain of MRP regulates binding to factor H,fibronectin,immunoglobulin G,and fibrinogen,and thus is the primary component in determiningS.suispathogenicity [10].MRP has been found to improve the viability ofS.suisand is implicated in the development of meningitis[12].Additionally,an MRP knockout strain showed an absence of adhesion ability to human brain microvascular epithelial cells[13].These evidence provides insight into a novel therapy forS.suis-associated diseases targeting the MRP-mediated pathogenicity ofS.suis.
The high mortality rate of inflammatory diseases evoked by pathogen infection is recognized to be attributable to abnormal immune responses,particularly in the form of inflammatory cytokine storms[14].While at the location of stimulation,immune cells including macrophages and neutrophils rely on their pattern recognition receptors (PRRs) to recognize pathogen-associated molecular patterns(PAMPs)and damage-associated molecular patterns.Toll-like receptor 2(TLR2)and TLR4 are two of the most well-studied PRRs because of their remarkable capacity to identify distinct molecular patterns displayed by invading pathogens.Despite the evidence that TLR2 and TLR4 are implicated in the recognition ofS.suisand activate the nuclear factor kappa B (NF-κB) signaling pathway via myeloid differentiation primary response 88 (MyD88) [15],it is not yet evident whether MRP is engaged in mediating the activation of TLR2 and TLR4 signaling in response toS.suis.
Recent years have noticed a rise in clinical data supporting the application of artemisinin and its derivatives as effective antimalarial medications.Artemisinins have also been proven to have antitumor,antifibrotic,antiviral,and antifungal properties.In mice infected with Acanthamoeba,artemisinin was found to lower TLR2 expression and alter TLR4 expression in the brain[16].Artemisinin's anti-inflammatory effects are due in part to a metabolite called dihydroartemisinin (DhA) [17].DhA suppressed TLR4 expression,which in turn inhibited type I interferon (IFN) expression and IFN regulatory factor 3 activation in mouse spleen cells challenged with lipopolysaccharides [18].Likewise,DhA alleviated the innate immune response induced by heat-inactivatedEscherichia coli(E.coli)in septicemic mice by inhibiting TLR4 transcription[19].In addition,the proinflammatory NF-κB and mitogen-activated protein kinase(MAPK) cascades are both downregulated by DhA,suggesting that this compound limits inflammatory insults [20-22].To combatS.suisinfection,it is essential to determine whether DhA is capable of mitigatingS.suis-initiated inflammatory response,with a focus on the key cytopathic effect mediated by MRP.
Here,MRP was cloned and expressed in vitro to analyze its involvement and pathomechanism inS.suis-induced inflammation in mice and macrophages.Given the underlying suppressive effect of DhA on inflammatory insults,we evaluated the efficacy of DhA in both in vitro and in vivo models for alleviating the MRP-initiated inflammatory insults.
2.Materials and methods
2.1.Reagents
We purchased theS.suisstrain CVCC 3913 from the China Veterinary Culture Collection Center.BD Biosciences (Franklin Lakes,NJ,USA) provided the brain heart infusion (BHI) solution.Yeast extract,agar powder,and tryptone used in this experiment were from Sangon Biotech (Shanghai,China).Dulbecco's modified Eagle's medium (DMEM),Lipofectamine 2000,and 0.25% trypsin/ethylene diamine tetraacetic acid (EDTA) were from Gibco BRL(Grand Island,NY,USA).HyClone Laboratories Inc.(South Logan,UT,USA)was contacted to acquire fetal bovine serum(FBS).Takara Biotechnology (Dalian) Co.,Ltd.(Dalian,China) supplied the Xho1 and Nde1 restriction enzymes.C29 and TAK-242 were obtained from MedChemExpress (Monmouth Junction,NJ,USA).The NF-κB inhibitor Bay11-7085 was provided by Sigma Chemicals Co.,Ltd.(St.Louis,MO,USA).S3I-201,an inhibitor targeting the activity of signal transducer and activator of transcription 3 (STAT3),was procured from Abcam (Cambridge,UK).TRIzol RNA extraction reagent was sourced from Aidlab Biotech Co.,Ltd.(Beijing,China).Complementary DNA(cDNA)Synthesis SuperMix,SYBR Green Master Mix,Zero-TOPO-Blunt Simple Cloning Kit,and isopropyl-β-D-thiogalactopyranoside (IPTG) were from Yeasen Biotech Co.,Ltd.(Shanghai,China).From Biolegend Co.,Ltd.(San Diego,CA,USA),we bought a LEGENDplex®enzyme-linked immunosorbent assay(ELISA) Kit.Antibodies against CD45,CD3,CD19,CD11b,CD64,Ly6G,NK1.1,CD68,and Gr-1 were from Biolegend Co.,Ltd.The phospho (p)-NF-κB p65 (Ser536) antibody and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody were procured from Santa Cruz Biotechnology,Inc.(Santa Cruz,CA,USA).From Cell Signaling Technology (Beverly,MA,USA),specific antibodies for phospho-STAT3 (Ser727),phospho-STAT3 (Tyr705),and STAT3 were obtained.Antibodies against TLR2,TLR4,MyD88,Janus kinase 2(JAK2),NF-κB p65,Toll-interleukin-1 receptor domain-containing adaptor protein,p-p38(Thr180/Tyr182),p-c-Jun N-terminal kinase(p-JNK) (Thr183/Tyr185),p-extracellular signal-regulated kinase(p-ERK) (Thr202/Tyr204),p38,JNK,and ERK were purchased from Sangon Biotech.Anti-p-JAK2 (Tyr1007/1008) were from Annoron Biotech Co.,Ltd.(Beijing,China).Antibodies produced from Huaxingbio Gene Technology Co.,Ltd.(Beijing,China),including Cy3-conjugated goat anti-rabbit secondary and peroxidase-conjugated goat anti-rabbit or goat anti-mouse,were purchased.Unless otherwise specified,all other chemicals,including DhA,were procured from Sigma-Aldrich (St.Louis,MO,USA).
2.2.Ethics statement
The Animal Ethics Committee of China Agricultural University approved all animal experiments (Approval No.: AW10180202-1-2).The experiments were conducted in accordance with the Guide for the Care and Use of Laboratory Animals,which was published by the Chinese Association for Laboratory Animal Science and Use.
2.3.Experimental animals
Fifty-four female C57BL/6 mice,aged 6 weeks,were obtained from Huafukang Biotechnology Ltd.(Beijing,China).For one week,the mice were kept in a 22-25°C,12 h light/dark cycle room with free access to food pellets (Huafukang Biotechnology Ltd.) and water until their body weights reached 21-25 g.To evaluate the inflammatory response and signaling triggered by MRP,30 mice divided into five groups were subjected to intraperitoneal injections of MRP (100 μL) dissolved in phosphate-buffered saline(PBS) at 0,0.25,0.5,1,or 2.5 mg/kg body weight.The mice were given MRP for 12 h after a 24 h fast and then sacrificed.To examine the ameliorating effect of DhA on MRP-induced inflammatory insults,24 mice randomly assigned to four groups were treated with either vehicle(olive oil)(control and MRP groups)or with 5 mg/kg body weight(i.p.)of DhA(DhA and DhA +MRP groups)once daily for six consecutive days,after which they fasted for one day and were exposed to PBS (vehicle) (control and DhA groups) or to 2.5 mg/kg body weight of MRP (MRP and DhA +MRP groups) for 12 h before sampling.Blood samples were taken by retroorbital puncture prior to sacrifice of the mice by cervical dislocation.Tissues from the spleens were either frozen in liquid nitrogen or fixed in optimal cutting temperature (OCT) and kept at -80°C for 24 h.
2.4.Cell culture and treatment
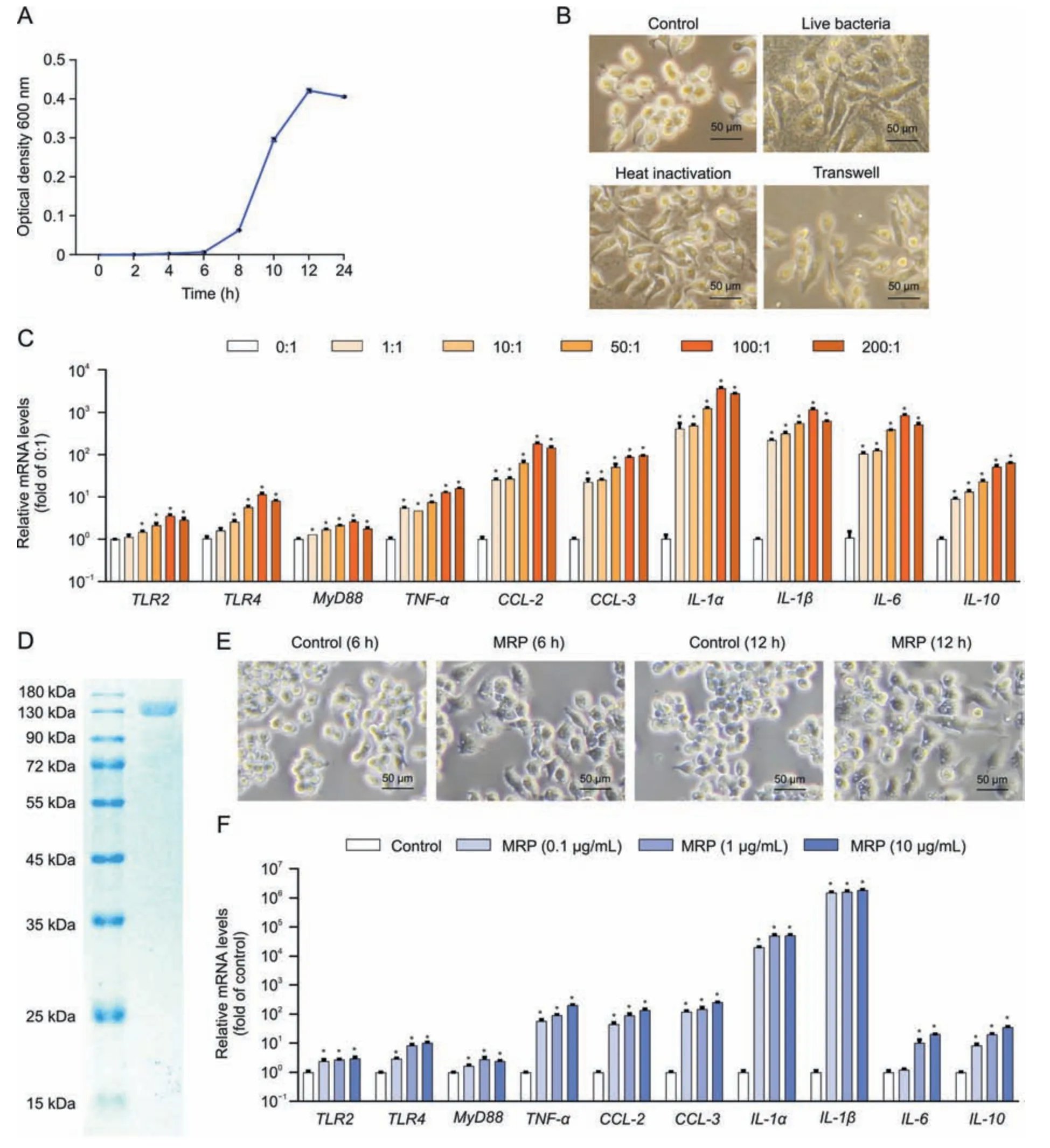
Fig.1.Muramidase-released protein(MRP)cloning and verification of its proinflammatory activity.(A)Streptococcus suis(S.suis)was cultured in Brain Heart infusion medium for 0,2,4,6,8,10,12,and 24 h before being harvested for absorbance detection (optical density 600 nm).(B) Alterations in RAW264.7 macrophage morphology induced by S.suis after 12 h of exposure to the pathogen in live,heat-killed,and Transwell conditions.The images were acquired by using a phase contrast microscope.(C)Messenger RNA(mRNA)levels of inflammation-related genes in S.suis-treated cells.RAW264.7 cells were incubated with the indicated multiplicity of infection of S.suis for 12 h,followed by mRNA determination by quantitative real-time polymerase chain reaction (PCR).Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal reference.(D) MRP was expressed in Escherichia coli (E.coli) by the pET26b (+) vector and purified by nickel-nitriloacetic acid affinity purification and ion exchange chromatography prior to analysis with sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Coomassie blue staining.(E) Morphological changes in RAW264.7 macrophages induced by MRP (10 μg/L)for 12 h.Images were acquired by using a phase contrast microscope.(F) mRNA levels of inflammation-regulatory genes in RAW264.7 cells exposed to 0,0.1,1,and 10 μg/mL MRP.GAPDH was used as an internal reference.Data are presented as the mean ± standard error of the mean (n =3).*P <0.05 vs.control. TLR2: Toll-like receptor 2; MyD88: myeloid differentiation primary response 88; TNF-α: tumor necrosis factor-α; CCL-2: chemokine C-C motif ligand 2; IL-1α: interleukin-1α.
RAW264.7 macrophages were grown in DMEM media containing 100 units/mL penicillin,100 μg/mL streptomycin,and 10%(V/V)FBS in a 37°C humidified incubator holding 5%CO2.Six-well plates were seeded with 5 × 105cells per well.Upon reaching 60%confluence,the cells were exposed toS.suis,MRP,DhA,or inhibitors in DMEM devoid of serum and penicillin-streptomycin.Next,the phenotypic changes of the cells were examined under a phasecontrast microscope(Olympus,Tokyo,Japan)with brightfield.
2.5.Bacterial strains, plasmids, and growth conditions
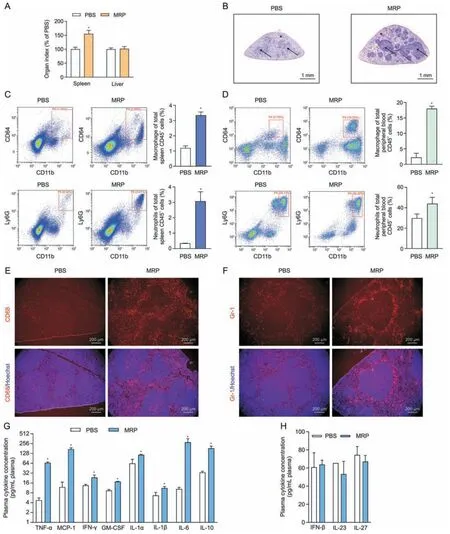
Fig.2.Treatment with muramidase-released protein (MRP) leads to infiltration of inflammatory cells and increased levels of inflammatory factors in mice.(A) The effect of the virulence protein MRP on the organ indices of the spleen and liver of mice.Organ index =organ weight/body weight.The dose of MRP for intraperitoneal injection was 2.5 mg/kg body weight in phosphate-buffered saline(PBS).(B)The results from hematoxylin&eosin(H&E)staining demonstrated the impact of MRP on the pathological change of the spleen from mice exposed to MRP.Asterisks point to red pulp,and arrows indicate white pulp.(C,D) Flow cytometry analysis of the effect of MRP on the number of white blood cell subtypes in the spleen(C)and plasma(D)of mice in response to MRP.CD64+CD11b+and Ly6G+CD11b+represent macrophages and neutrophils,respectively;the corresponding bar graph on the right side of the scatter plot represents the percentage of cells from each group of 6 mice.(E,F) The effect of MRP on the number and distribution of mouse spleen macrophages (E) and neutrophils (F).CD68 and Gr-1 are used as the surface markers of macrophages and neutrophils,respectively.The cell nucleus was stained using Hoechst 33,342 (5 μg/mL).(G) The levels of plasma inflammatory cytokines that exhibited a significant increased in response to MRP.(H) The concentrations of interferon-β (IFN-β),interleukin-23(IL-23),and IL-27 following PBS or MRP treatment.Data are expressed as the mean±standard error(n =6).*P <0.05 vs.PBS(control).TNF-α:tumor necrosis factorα;MCP-1: monocyte chemoattractant protein-1;GM-CSF: granulocyte macrophage colony-stimulating factor.

Fig.3.Regulation of inflammation-related signal pathways by muramidase-released protein(MRP)in the spleen of mice.Mice were exposed to MRP treatment with the indicated doses for 12 h.(A,C) The effect of MRP on the expression of pattern recognition receptor (PRR)-related proteins.(B,D) MRP drives the phosphorylation of proteins in the nuclear factor kappa B (NF-κB) and mitogen-activated protein kinase (MAPK) signaling pathways.The representative bands shown are the protein or phosphorylated protein levels in mouse spleen detected by Western blot.Gray values for each group were measured with ImageJ 1.8.0 and displayed as bar graphs.Values were normalized to the expression of glyceraldehyde-3-phosphate dehydrogenase(GAPDH)or the total protein corresponding to each phosphorylated protein.Data are expressed as the mean±standard error(n =6).*P <0.05 vs.0 mg/kg body weight group.TLR2: Toll-like receptor 2;MyD88: myeloid differentiation primary response 88;TIRAP: Toll-interleukin-1 receptor domain-containing adaptor protein;NF-κB: nuclear factor kappa B;JAK2: Janus kinase 2;STAT3: signal transducer and activator of transcription 3;JNK: c-Jun N-terminal kinase;ERK: extracellular signal-regulated kinase.
Before being collected through centrifugation in the late exponential expansion stage,S.suisstrain CVCC 3913 was grown at 37°C in BHI medium (pH 7.4) or on BHI agar plates with a pH of 7.4.At 37°C,Luria-Bertani broth or agar plates were used for the proliferation ofE.coli TOP10andRosetta-gami(DE3)pLysSstrains.The pESI-Blunt and pET-26b(+)vectors were used for assisting protein expression.
2.6.Real-time polymerase chain reaction (PCR) assay
Following the directions provided by the manufacturer,a TRIzol reagent was applied for the purpose to separate total RNA from either the spleens of mice or RAW264.7 cells.The isolated RNA was stored in freezers at -80°C prior to proceeding with reverse transcription.The steps below were used to obtain cDNA using the Hifair®II SuperMix Plus reagent,per the procedure as follows:25°C for 5 min;42°C for 30 min;and 85°C for 5 min.After combining the Hieff®quantitative PCR SYBR Green Master Mix reagent,upstream and downstream primers,and cDNA samples,PCR amplifications were performed in accordance with the guidelines included in a kit using an ABI 7500 real-time quantitative PCR instrument (Life Technologies,Carlsbad,CA,USA).The following is an outline of the steps that were taken during the reaction:1 cycle of 5 min at 95°C,followed by 10 s at 95°C and 34 s at 60°C for a total of 40 cycles.Target gene expression was quantified in comparison to the housekeeping gene GAPDH using the 2-ΔΔCTmethod[23].Table S1 displays the primer sequences for the real-time PCR assay.
2.7.Expression and purification of MRP
Through the use of the primers (MRP forward primer: CGCCATATGATGGGTGCTGGTGCACA;MRP reverse primer: CTCGAGATCTTCGTTACGACGAC;product size: 3600-3700 bp) and KAPA Hifi DNA polymerase (Kapa Biosystems,Wilmington,MA,USA),E.coliwas genetically modified to express MRP by performing PCRonS.suisgenomic DNA.The expression method was carried out in accordance with the procedure described previously [24].The PCR products were cloned into pET-26b(+)vectors andthentransformed into Rosetta-gami (DE3) pLysS strains.Verification of cloned sequences was achieved through DNA sequencing (T7 forward sequencing primer: TAATACGACTCACTATAGGG;T7 reverse sequencing primer: TGCTAGTTATTGCTCAGCGG).To purify the protein,we used nickel-chelating chromatography (GE Healthcare,Upsala,Sweden) and ¨AKTA ion exchange chromatography (GE Healthcare) following the instructions provided by the manufacturers.Briefly,following centrifugation and washing,the bacteria were resuspended in lysis buffercontaining 200μg/mL lysozyme and 1 mM bemethyl sulfonyl fluoride,andthe suspensionwas clarified by ultrasonication at 4°C.After centrifugation and filtration,the target protein was purified by a nickel column (nickel-nitriloacetic acid)and concentrated to~1 mg/mL by an ultrafiltration tube.The protein solution obtained was purified by ion exchange chromatography according to theinstructions of¨AKAT ion-exchange chromatography.Thereafter,the corresponding effluent of all ultraviolet absorption peaks was collected.Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Coomassie brilliant blue staining were carried out to examine the effluent.After desalting the target protein,the concentration was adjusted to 1 mg/mL.The purified protein was stored at-80°C.Target protein was desalted,and then diluted to 1 mg/mL.Protein after purification was frozen at-80°C.
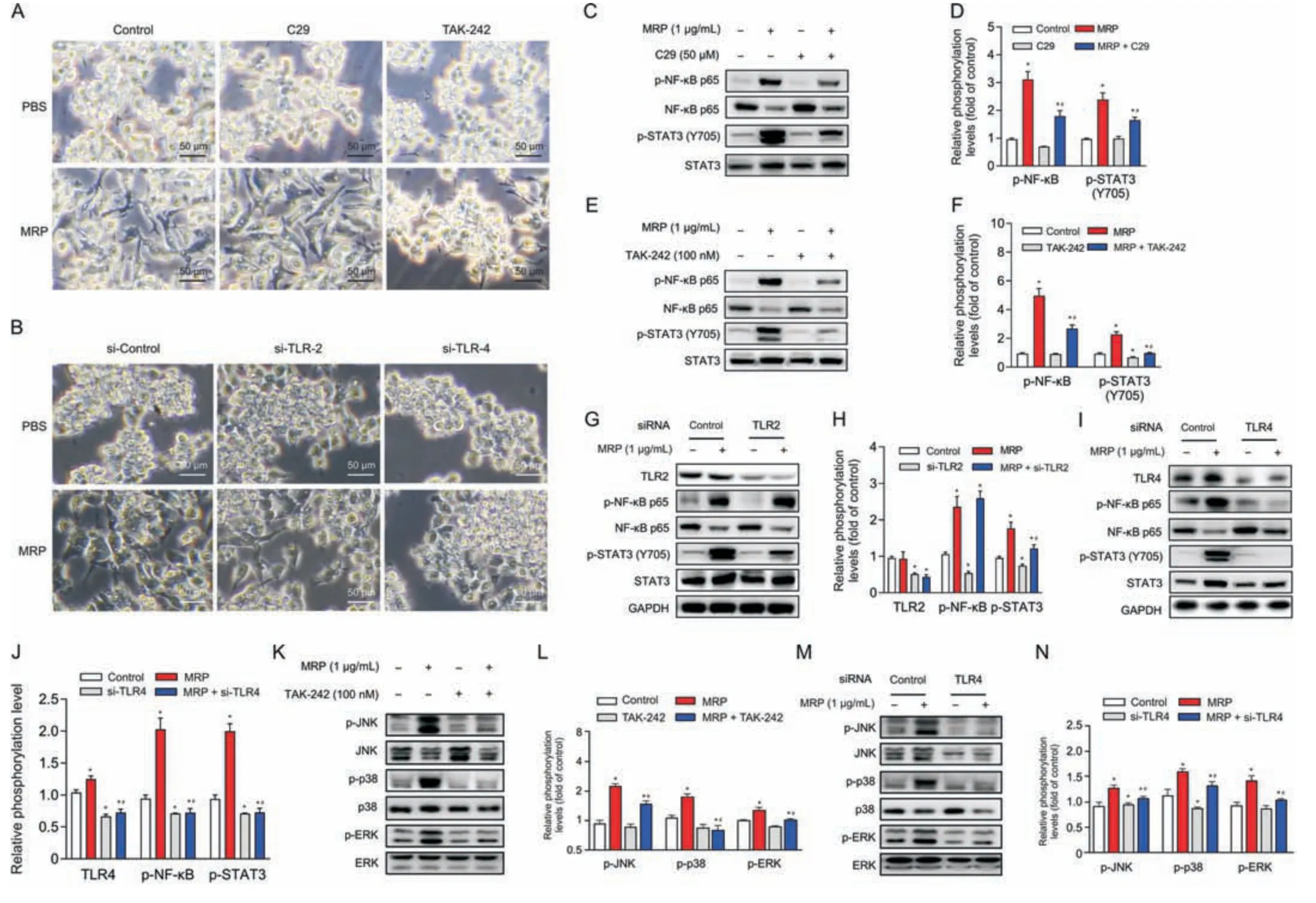
Fig.4.Toll-like receptor 4 (TLR4) serves as a pivotal receptor mediating the inflammatory response induced by muramidase-released protein (MRP).(A,B) Morphological alterations in RAW264.7 cells following TLR2 or TLR4 intervention with inhibitor(C29 or TAK-242)or small interfering RNA(siRNA).(C-N)Immunoblot analysis of proteins associated with inflammation.All band intensities were quantified by ImageJ (version 1.8.0).The corresponding nonphosphorylated proteins or glyceraldehyde-3-phosphate dehydrogenase(GAPDH)were regarded as loading references.The results are reported as the mean±standard error(n =3).*P <0.05 vs.control.#P <0.05 vs.MRP.PBS:phosphate-buffered saline;NF-κB: nuclear factor kappa B;STAT3: signal transducer and activator of transcription 3;JNK: c-Jun N-terminal kinase;ERK: extracellular signal-regulated kinase.
2.8.Histopathological examination and immunohistochemistry
The tissue samples from the spleen were frozen at-80°C for 24 h in OCT.Staining with hematoxylin and eosin(H&E)was performed on tissue slices cut from the spleens at a thickness of 5 μm using a portable frozen microtome(Thermo Fisher Scientific Inc.,San Jose,CA,USA) to evaluate the extent of inflammatory cell infiltration.Tissue sections were exposed to heat treatment to facilitate antigen retrieval and then blocked with 1%goat serum for 10 min at 25°C before being exposed to an incubation with a primary antibody against CD68 or GR-1 at 4°C for 16 h.Thereafter,following three washes in PBS,the sections were put into an incubation solution containing Cy3-conjugated secondary antibodies(1:100)for 1 h at 25°C.The nuclei were stained for 10 min at 25°C with 5 μg/mL Hoechst 33,342.Distributions of individual proteins were identified using a fluorescence microscope(Axio Vert.A1;Zeiss,Jena,Germany).
2.9.Western blot analysis
Proteins isolated from cells or tissues were separated(25 μg for each sample) using SDS-PAGE gels and then transferred to polyvinylidene difluoride membranes (Millipore,Billerica,MA,USA).Following a 24 h incubation at 4°C,the primary antibody(1:1000)was added to each membrane that had been blocked with 5%nonfat milk.Subsequently,a 1:2000 dilution of an HRPconjugated secondary antibody was then employed to incubate the membranes for 1 h at 25°C.Following incubation with an enhanced chemiluminescence plus reagent,blots were monitored by an Image Quant LAS 4000 mini system (GE Healthcare).Using ImageJ software,chemifluorescence was measured (Bio-Rad Laboratories,Hercules,CA,USA).All of our data were normalized to either GAPDH or the corresponding nonphosphorylated protein,and the results were given as a percentage in relation to the control group.
2.10.Immunofluorescence assay for cultured cells
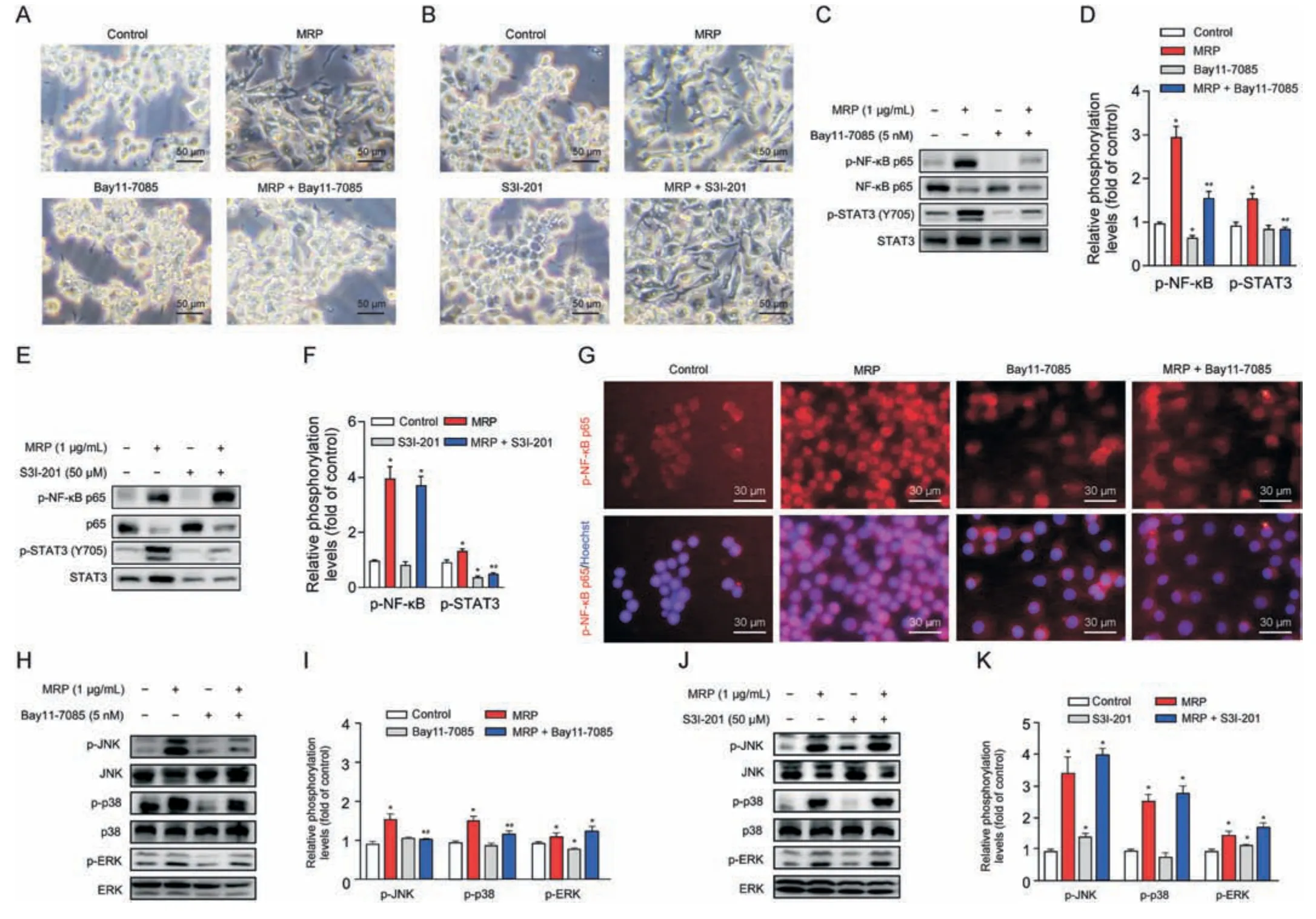
Fig.5.Signaling pathways downstream of nuclear factor kappa B(NF-κB)are involved in the mechanism concerning muramidase-released protein(MRP)-disturbed macrophages.(A,B)Microscopic assessment of morphological alterations in RAW264.7 cells subjected to Bay11-7085(A)or S3I-201(B)pretreatement followed by MRP exposure.(C-F)Western blot analysis of NF-κB p65 and signal transducer and activator of transcription 3 (STAT3) phosphorylation.(G) Immunofluorescence detection of p-NF-κB.(H-K) Western blot analysis of mitogen-activated protein kinases (MAPKs) (c-Jun N-terminal kinase (JNK),p38,and extracellular signal-regulated kinase (ERK)) phosphorylation.Quantification of bands with reference to the corresponding nonphosphorylated protein was analyzed by ImageJ software (version 1.8.0).The results are expressed as the mean ± standard error(n =3).*P <0.05 vs.control.#P <0.05 vs.MRP.
Using Triton X-100 diluted to 0.1%(V/V)in PBS,RAW264.7 cells were permeabilized at 4°C for 10 min,after being exposed to 4%paraformaldehyde(fixation solution)for 20 min at 25°C.Following blocking for 1 h at 25°C in 1%goat serum in PBS,the samples were exposed to primary antibody targeting NF-κB p65 (1:200) for 16 h at 4°C.Thereafter,the samples were subjected to Cy3-conjugated secondary antibodies (1:100) for 1 h at 25°C after being washed three times with PBS.Hoechst 33,342 (5 μg/mL) staining was performed to discriminate the nuclei of the cells for 10 min at 25°C.A fluorescence microscope was employed for observation and image capture (Axio Vert.A1;Zeiss,Germany).
2.11.Statistical analysis
The mean is followed by the standard error of the mean when statistics are presented.To analyze the data and its significance,GraphPad Prism (version 9) was used to perform a one-way analysis of variance or a Student's two-tailedttest with a Student-Newman-Keuls multiple comparison.Our criterion for evaluating the significance between groups was to observe whether thePvalue was less than 0.05.
3.Results
3.1.MRP cloning and its pro-inflammatory activity verification
As shown in Fig.1A,S.suisexhibited a typical S-shaped growth curve within 24 h in BHI medium.RAW264.7 cells were treated with live,heat-killed,or Transwell separatedS.suisat a multiplicity of infection of 200 for 24 h.We observed that both live and heatkilled bacteria resulted in macrophage polarization,and thatS.suisalso led to polarization of RAW264.7 cells under noncontact conditions by using Transwells (Fig.1B).Macrophage M1-type polarization is commonly accompanied by proinflammatory response.S.suismarkedly increased the messenger RNA (mRNA)abundances of genes pertaining to PRRs (TLR2,TLR4,andMyD88),cytokinestumor necrosis factor-α(TNF-α),interleukin-1α(IL-1α),IL-1β,IL-6,andIL-10,and chemokines (chemokine C-C motif ligand 2(CCL2)andCCL-3)(P<0.05)(Fig.1C).These findings prompted us to postulate that theS.suisstrain used in this study is pathogenic and that its virulence factor may play a role in the occurrence of the inflammatory process.Next,we cloned and purified MRP as shown in Fig.1D.To verify the activity of MRP,macrophages were treated with 10 μg/mL MRP.The cells exhibited M1 polarization at 6 h and 12 h in response to MRP treatment (Fig.1E).In addition,MRP markedly elevated the mRNA levels of inflammation-related genes,includingTLR2,TLR4,TNF-α,MyD88,IL-6,IL-10,IL-1α,IL-1β,CCL-2,andCCL-3in a dose-dependent manner (P< 0.05) (Fig.1F).Following the above results,the virulence protein MRP ofS.suisshares the same proinflammatory properties as liveS.suisin macrophages,suggesting that it may be a key determinant of the proinflammatory response ofS.suis.
3.2.MRP leads to inflammatory conditions in the spleen and blood of mice
Mice were received injections intraperitoneally with MRP(2.5 mg/kg body weight) and then were slaughtered after a 12 h exposure in order to explore the proinflammatory activity of MRP in vivo.As shown in Fig.2A,the organ index(organ weight to body weight ratio) of the spleen rather than the liver was significantly increased by MRP (P<0.05).Histological sectioning showed an increase in leukocyte infiltration in the white pulp of the spleen in MRP-treated mice (Fig.2B).Flow cytometry and immunohistochemistry were conducted to identify changes in macrophages and neutrophils induced by MRP.We observed that MRP significantly increased the percentage of macrophages(CD64+and CD11b+)and neutrophils (Ly6G+and CD11b+) in the spleen (Fig.2C) and peripheral blood (Fig.2D) (P<0.05).Next,immunofluorescence was performed to assess the accumulation of macrophages and neutrophils in spleen tissues.Compared with the controls,MRP treatment dramatically increased the number of CD68+and Gr-1+cells in the spleen (P<0.05) (Figs.2E and F).Additionally,mice challenged with MRP exhibited higher levels of plasma IL-1α,IL-1β,IL-6,IL-10,TNF-α,IFN-γ,monocyte chemoattractant protein-1 (MCP-1),and granulocyte-macrophage colony-stimulating factor (GMCSF) in comparison with the mice from control group (P<0.05)(Fig.2G).However,MRP showed no noticeable impact on the plasma concentrations of IL-23,IL-27,and IFN-β in mice challenged with MRP (Fig.2H).
3.3.MRP activates TLR4, NF-κB, MAPKs, and JAK2-STAT3 signaling in mouse spleen
The initiation of inflammation concerns the recognition of the pathogenic antigen by PRRs.TLR4 and MyD88 expression levels were significantly (P<0.05) upregulated in MRP-treated mouse spleen tissue,while TLR2 expression was unaffected (Figs.3A and C).Moreover,MRP enhanced the phosphorylation levels of NF-κB,JAK2,STAT3,and MAPKs(JNK,p38,and ERK)in mouse spleens in a dose-dependent manner (P<0.05) (Figs.3B and D).
3.4.The inflammatory response induced by MRP relies on TLR4-dependent activation of NF-κB p65, STAT3, and MAPKs
To determine whether or not TLR2 and TLR4 are implicated in MRP-triggered inflammatory signaling in RAW264.7 macrophages,C29 (a TLR2 inhibitor) and TAK-242 (a TLR4 inhibitor) were supplemented to the medium prior to MRP exposure.Cells were pretreated with C29 or TAK-242 for 6 h,followed by MRP exposure for 12 h.TAK-242 obviously inhibited MRP-induced macrophage M1 polarization,as shown in Fig.4A,whereas C29 pretreatment showed no effect on the morphological change.However,both inhibitors exhibited inhibitory effects on the MRP-initiated NF-κB p65 and STAT3 phosphorylation (P< 0.05) (Figs.4B-E).The involvement of TLR2 and TLR4 in MRP-treated macrophages was further validated by RNA interference.As expected,similar outcomes were observed in cells subjected to small interfering RNA (siRNA) targeting TLR4 (Figs.4F,I,and J).Knockdown of TLR2 by siRNA,however,did not ameliorate the phosphorylation of NF-κB p65 induced by MRP (Figs.4G and H).Likewise,inhibition or knockdown of TLR2 showed no effect on MAPK phosphorylation (p-JNK,p-p38,and p-ERK) (data not shown),whereas TLR4 intervention significantly suppressed p-JNK,p-p38,and p-ERK levels in MRP-challenged cells (Figs.4K-N).Therefore,we deduced that TLR4-governed NF-κB p65,STAT3,and MAPKs played a crucial role in the initiation of inflammatory signaling.
3.5.NF-κB, which is upstream of STAT3 and MAPKs, is involved in the MRP-induced proinflammatory response
Next,we further characterized the interactions between NF-κB and STAT3 or MAPKs in MRP-treated macrophages.The NF-κB p65 inhibitor Bay11-7085 and the STAT3 inhibitor S3I-201 were added prior to MRP exposure.Cells were pretreated with Bay11-7085 or S3I-201 for 2 h and then subjected to MRP for 12 h.Bay11-7085 significantly inhibited MRP-induced macrophage polarization,whereas S3I-201 presented no discernible effect (Figs.5A and B).Western blot data demonstrated that Bay11-7085 markedly reduced the phosphorylation of NF-κB p65 and STAT3 induced by MRP,and also suppressed the phosphorylation levels of p38 and JNK (P<0.05) (Figs.5C,D,H and I).However,the STAT3 inhibitor S3I-201 showed no obvious effect on the phosphorylation of NF-κB p65 and MAPKs,although it presented inhibitory effects on STAT3 activity (Figs.5E,F,J,and K).In addition,MRP induced the translocation of NF-κB p65 into the nucleus from the cytoplasm,which was inhibited by Bay11-7085 (Fig.5G).The above data indicated that the activity of MAPK and STAT3 signaling is modulated by NFκB in cells subjected to MRP challenge,which in turn leads to a proinflammatory phenotype.
3.6.DhA relieves MRP-induced inflammatory insults
DhA is known as a derivative of artemisinin that possesses antiinflammatory properties.Therefore,the contribution of DhA to protection against MRP toxicity in RAW264.7 macrophages and mice was examined.Pretreatment with DhA for 6 h displayed an obvious inhibitory effect on macrophage M1 polarization induced by MRP (Fig.6A).The increase in the mRNA levels ofTLR4,TNF-α,MyD88,IL-6,IL-10,IL-1α,IL-1β,andCCL-2evoked by MRP in RAW264.7 cells was effectively attenuated in response to DhA treatment (P<0.05) (Fig.6B).Treatment of cells with DhA significantly attenuated the activation of NF-κB p65,MAPKs (JNK,p38,and ERK),and JAK2-STAT3 induced by MRP,as well as the protein levels of TLR4 and MyD88 (Figs.6C-H).In vivo,the extensive infiltration of inflammatory cells in the spleen of MRP-challenged mice was greatly ameliorated by DhA (Fig.7A).The results of flow cytometry indicated that DhA alleviated the elevation in the proportion of macrophages and neutrophils in both spleen tissue and peripheral blood of mice exposed to MRP (P<0.05) (Figs.7B-E).Moreover,DhA suppressed the plasma concentrations of IL-1α,IL-1β,IL-6,TNF-α,MCP-1,and GM-CSF in mice challenged with MRP(P<0.05) (Figs.7F and G).When compared to the MRP-treated group,the combination of DhA and MRP injections into mice resulted in a significant reduction in the level of TLR4;however,the injections had no effect on the levels of TLR2 or MyD88 (Fig.8A).Additionally,DhA inhibited the of NF-κB,JAK2-STAT3,and MAPK(JNK and ERK) activation triggered by MRP in mouse spleen (Figs.8B-D).Taken together,these observations indicate that DhA protects against MRP-induced inflammation by blocking TLR4-governed activation of NF-κB followed by STAT3 and MAPKs signaling.
4.Discussion
The bacteriumS.suishas emerged as a pernicious pathogen responsible for a wide range of illnesses,including septic shock,polyserositis,meningitis,and even sudden death.To date,the pathogenesis of infectious diseases elicited byS.suishas not been adequately addressed.However,the virulence factor MRP expressed byS.suishas been identified as one of the key determinants mediating the severity and progression ofS.suisassociated diseases.Elucidating the mechanism concerning MRP-mediated inflammation will provide intervention targets forS.suisinfection.In the present study,we demonstrate that TLR4-dependent activation of NF-κB-STAT3/MAPK signaling is required for MRP-induced proinflammatory response.Based on these targets,we found that DhA,a derivative of artemisinin,has the capacity of to relieve the MRP-induced inflammatory response.
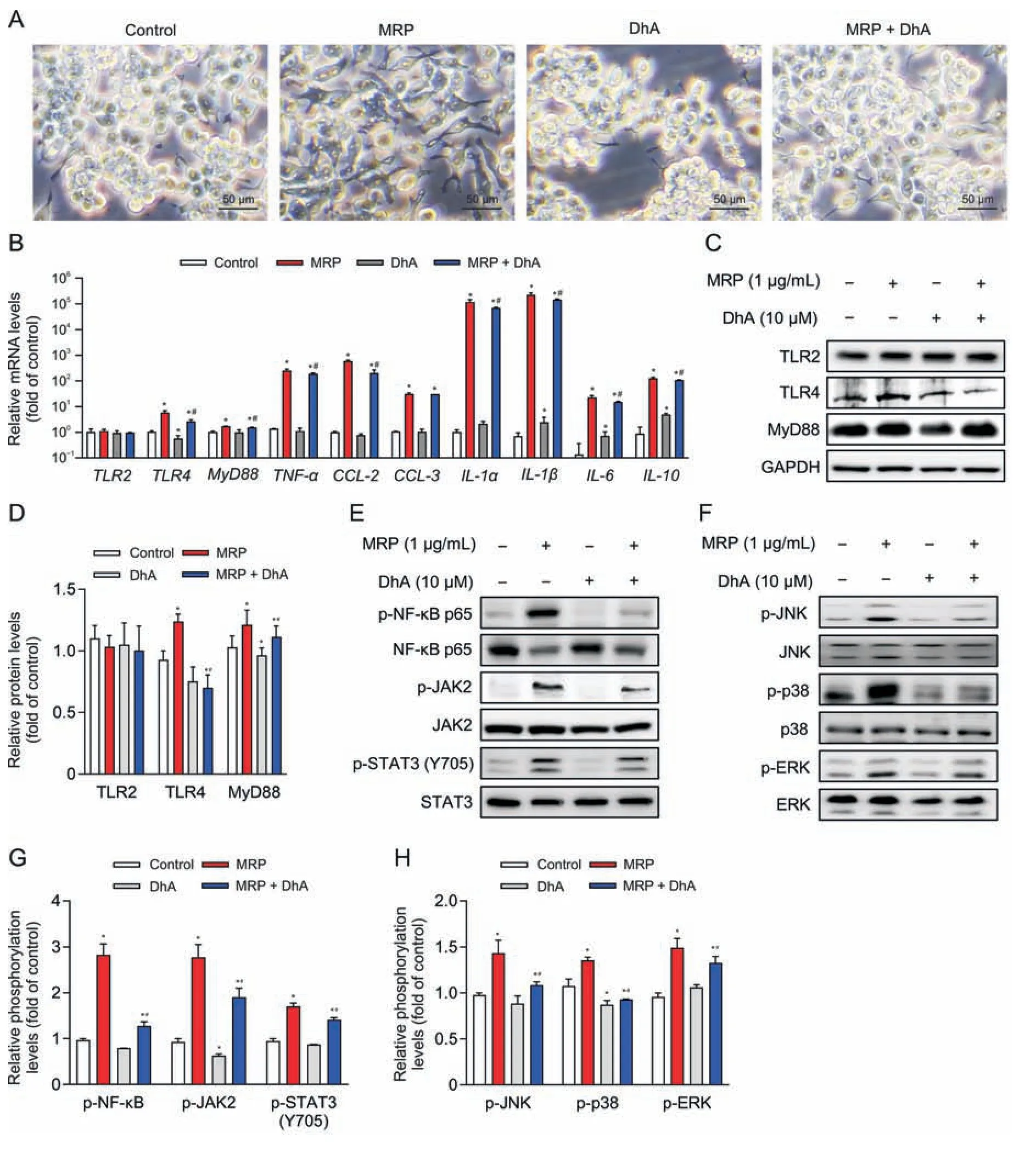
Fig.6.Dihydroartemisinin (DhA) relieves the muramidase-released protein (MRP)-induced inflammatory response in RAW264.7 cells.(A) Microscopic observation of cells pretreated with DhA (10 μM) followed by MRP (1 μg/mL) exposure.(B) Quantitative real-time polymerase chain reaction (PCR) analysis of the expression of inflammatory factors.Glyceraldehyde-3-phosphate dehydrogenase(GAPDH)was considered as an internal control gene.(C-H)Western blot analysis of protein abundances and phosphorylation levels.The band intensities representing protein expression or phosphorylation level were quantitated with reference to GAPDH or total protein control bands using ImageJ 1.8.0) (n =3).*P <0.05 vs.control.#P <0.05 vs.MRP.TLR2: Toll-like receptor 2;MyD88: myeloid differentiation primary response 88;TNF-α: tumor necrosis factor-α;CCL-2: chemokine C-C motif ligand 2;IL-1α:interleukin-1α;NF-κB:nuclear factor kappa B;JAK2: Janus kinase 2;STAT3:signal transducer and activator of transcription 3;JNK:c-Jun N-terminal kinase;ERK: extracellular signal-regulated kinase.
It has been widely accepted that M1-polarized macrophages act as vital modulators of infectious diseases and systemic inflammation.Macrophage M1 polarization occurs in response to external stimuli (e.g.,microorganisms or proinflammatory factors) and expresses proinflammatory factors and chemokines providing antigen-presenting function[25].In a mouse infection model,IL-1 production from bone marrow-derived macrophages has been implicated in bacterial clearance and systemic inflammation induced byS.suis[26],suggesting that macrophage polarization is indeed responsible for the pathomechanisms concerningS.suis.In vitro,S.suishas been reported to provoke a proinflammatory phenotype in macrophages [27].Consistently,we found that macrophages exhibited the phenotype of M1 polarization following the treatment of virulence factor MRP,and the expression of M1 polarization markers (TNF-α,CCL-2,IL-1,andIL-6) was markedly upregulated;these changes were similar to the observations in the canonical proinflammatory polarization model of macrophages induced by bacterial lipopolysaccharides [28],suggesting that the proinflammatory response triggered byS.suisis attributed,at least in part,to macrophage M1 polarization induced by MRP secreted byS.suis.
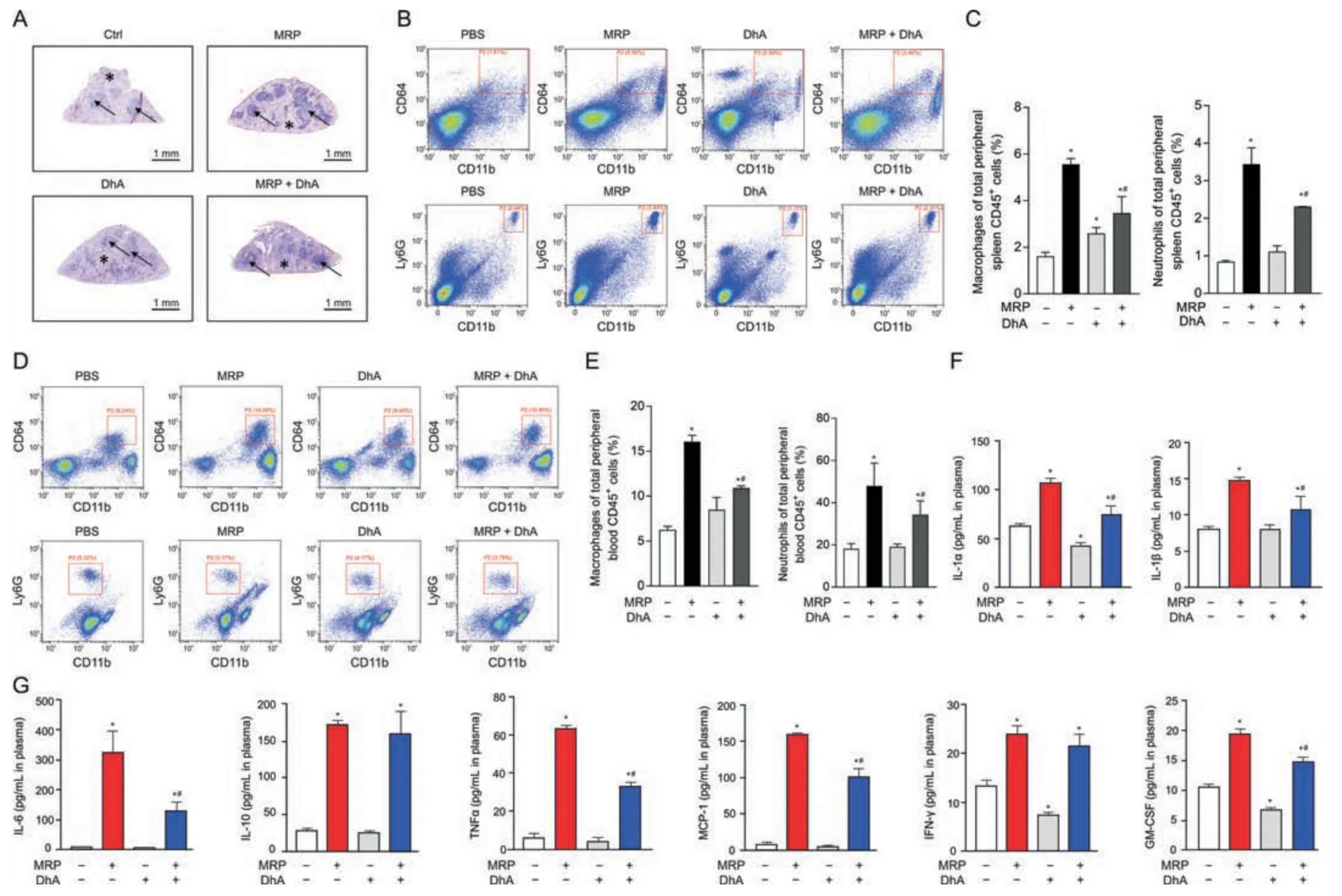
Fig.7.Dihydroartemisinin (DhA) mitigates muramidase-released protein (MRP)-induced splenomegaly and inflammatory response in mice.(A) Hematoxylin &eosin (H&E)staining of the spleen tissues.(B,C) Flow cytometry analysis of macrophages (CD64+CD11b+populations) and neutrophils (Ly6G+CD11b+populations) isolated from the spleens.Plasma samples were used for (D,E) CD64+CD11b+macrophage and Ly6G+CD11b+neutrophil analysis and (F,G) enzyme-linked immunosorbent assay (ELISA) for inflammatory cytokines.Data are expressed as the mean ± standard error (n =6).*P <0.05 vs.control.#P <0.05 vs.MRP.PBS: phosphate-buffered saline;IL-1α: interleukin-1α;TNF-α: tumor necrosis factor-α;MCP-1: monocyte chemoattractant protein-1;IFN-γ: interferon-γ;GM-CSF: granulocyte-macrophage colony-stimulating factor.
Inflammation induced by bacterial virulence factors is dependent on PRRs sensing PAMPs derived from pathogens.TLR2 and TLR4 serve as crucial and widely studied PRRs mediating the activation of innate immunity.Graveline et al.[29] found that CPS derived fromS.suisactivated the macrophage inflammatory response through TLR2 and MyD88 instead of TLR4.TLR2 also participates inS.suis-induced activation of mouse astrocytes,splenic inflammation,and the macrophage immunological response [30-32].By comparison,compelling studies have indicated that TLR4-dependent pathways are key candidates involved in the immunological targets ofS.suisinfection [33,34].These evidence urge us to identify whether TLR2 and/or TLR4 mediates the proinflammatory response induced by MRP derived fromS.suis.Our data provide evidence that the inflammatory stress induced by MRP is linked with TLR4 in place of TLR2.TLR4,which initially emerged as a receptor activated by lipopolysaccharides,has been known to evoke cascades of inflammatory signals.Activation of TLR4 signaling is one of the main reasons for the progression of sepsis and other bacterial infectious diseases [35].TAK-242 (also known as resatorvid),a specific inhibitor of TLR4 signaling,effectively inhibited TLR4 ligand-dependent inflammatory signal transduction,thereby affecting the phosphorylation of the NF-κB cascade[36].In parallel to this work,we observed that inhibition of TLR4 led to a reduction in NF-κB p65 phosphorylation.
MAPKs and STAT3 are known as major signaling molecules modulating immune responses induced by a range of bacterial infections.Indeed,an infection withS.suiswas proven to actuate the MAPK and STAT3 signaling pathways [37,38].MAPK signaling has been identified as a target downstream of TLR4[39].Our study also provided evidence for TLR4-modulated MAPK activation in MRPtreated macrophages.In addition,we demonstrated that NF-κB governs the activity of STAT3 and MAPKs in macrophages exposed to MRP.In agreement with our results,NF-κB has been verified to be a master regulator of the activation of STAT3 phosphorylation in anHelicobacter pyloriinfection model [40].In addition,activation of MAPKs downstream of NF-κB was noted in lipopolysaccharidestreated macrophages [41].These evidence delineate the involvement of TLR4 in NF-κB activation and subsequent cascades of STAT3 or MAPKs in macrophages subjected to MRP treatment.
It has been proposed for years that artemisinin and its derivatives,such as DhA,possess anti-inflammatory and immunoregulatory properties,and more evidence is piling up to support this assertion.For example,DhA assists the proliferation of Th and Treg cells and thus the amelioration of inflammatory disease,which is associated with the activation of mTOR signaling [42].In addition,mice with inflammatory bowel disease treated with DhA exhibited an inhibition in CD4+T lymphocyte activation and a restoration in the balance of Th/Treg [43].A recent study of piglets with intrauterine growth retardation revealed that DhA mitigated the inflammatory response in the gut through TLR4-NOD-NF-κB signaling[44].Our results showed that DhA effectively inhibited MRPinduced macrophage accumulation and inflammation by blocking of TLR4-controlled activation of NF-κB-STAT3/MAPKs signaling.Consistent with our findings,the results from lipopolysaccharidechallenged RAW264.7 macrophages showed that artesunate,an artemisinin derivative,weakens proinflammatory responses via the inhibition of TLR4 [19].Evidence from microglial cells supports a link between TLR4 and MAPK signaling and the generation of proinflammatory cytokines in response to lipopolysaccharide stimulation,which were blocked by artemisinin [45].Taken together,these results point to the anti-inflammatory protective capacity and potential mechanisms of artemisinin and its derivatives in infectious diseases.
5.Conclusion
In conclusion,we shed light on a novel mechanism underlying the proinflammatory response in macrophages induced by MRP,which is primarily associated with TLR4-governed NF-κB activation followed by STAT3 and MAPK signaling.DhA offers protection against the inflammatory response initiated by MRP and may attenuate the clinical disease severity induced byS.suisinfection.Administration of DhA inhibited the proinflammatory response elicited by MRP via suppression of TLR4 protein expression and subsequent NF-κB phosphorylation followed by MAPK or JAK2-STAT3 activation.Our study not only revealed a signaling mechanism implicated in the inflammatory response induced by the virulence protein MRP but also unveiled the role of DhA in alleviating MRP-elicited inflammatory insults,providing a theoretical rationale for the application of DhA in the amelioration of inflammatory diseases induced byS.suis.
CRediT author statement
Yun Ji:Methodology,Writing -Original draft preparation,Reviewing and Editing,Funding acquisition;Kaiji Sun:Methodology,Investigation,Formal analysis,Writing -Original draft preparation;Ying Yang:Methodology,Formal analysis,Supervision,Writing-Reviewing and editing;Zhenlong Wu:Conceptualization,Funding acquisition.
Declaration of competing interest
The authors declare that there are no conflicts of interest.
Acknowledgments
This work was supported by the National Key R&D Program of China (Grant Nos.: 2022YFF1100104 and 2022YFF1100102),the National Natural Science Foundation of China (Grant Nos.:31625025,32172749,and 32202701),the 2115 Talent Development Program of China Agricultural University (Grant No.: 00109016),and the Zhengzhou 1125 Talent Program,China (Grant No.:2016XT016).
Appendix A.Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jpha.2023.05.013.
 Journal of Pharmaceutical Analysis2023年10期
Journal of Pharmaceutical Analysis2023年10期
- Journal of Pharmaceutical Analysis的其它文章
- Protective effects of catalpol on cardio-cerebrovascular diseases: A comprehensive review
- Recent advancements in single-cell metabolic analysis for pharmacological research
- SPME as a green sample-preparation technique for the monitoring of phytocannabinoids and endocannabinoids in complex matrices
- Monoclonal antibody targeting mu-opioid receptor attenuates morphine tolerance via enhancing morphine-induced receptor endocytosis
- Protective effects of dioscin against Parkinson's disease via regulating bile acid metabolism through remodeling gut microbiome/GLP-1 signaling
- Histone deacetylase inhibitor pracinostat suppresses colorectal cancer by inducing CDK5-Drp1 signaling-mediated peripheral mitofission