
GPR30受体在神经退行性疾病和脑缺血中的作用
2023-12-05 11:15:04潘彦灼吴凌智综述何玲审校
海南医学 2023年22期
潘彦灼,吴凌智 综述 何玲 审校
1.中国药科大学药学院,江苏 南京 210009;
2.嘉兴市第一医院,浙江 嘉兴 314000
GPR30 受体是一种七次跨膜GPCR,其于1997 年间被发现[1]。随后因观察到GPR30 受体可以与雌激素相互作用并且介导雌激素效应,GPR30 受体于2007 年被国际基础临床药理学协会正式命名为G 蛋白偶联雌激素受体(G protein coupled estrogen receptor,GPER)[2]。与其他雌激素受体不同的是,GPR30受体主要介导雌激素的非基因组效应[3]。该效应涉及的分子信号起始于膜上,终止于细胞质当中,整个过程发生迅速,使靶细胞能够在极短时间内响应雌激素,比如包括在数分钟内改变离子通道以及激酶信号通路强弱[4]。作为一种新型雌激素受体,其功能被广泛与雌激素已知效应比较。雌激素通常被认为一类由性腺分泌的类固醇激素,参与调控女性性发育以及生殖全过程。然而目前已经认识到雌激素具有广泛的生理作用,皮肤系统、中枢神经系统、心血管系统、骨骼系统、免疫系统、代谢系统和排泄系统都是其直接调控对象[5]。其中雌激素的神经作用催生出雌激素替代疗法[6-7],选择性靶向雌激素核受体[8]等策略用于治疗各种神经系统疾病。
随着全球多地区的老龄化程度加剧[9-10]以及不健康的生活习惯[11],神经退行性疾病以及心脑血管疾病呈现出大流行趋势。该类型流行将造成大量身心残障人群出现并进一步给患者,家庭,社会以及国家带来巨大压力。在神经退行性疾病临床治疗中还尚未出现十分有效的诊治管理策略,对症治疗策略的药效局限性[12],神经退行性疾病的集聚一身特性和占比异质性导致单一药物治疗这些患者成为奢望[9-10]。作为全球另一高发病率以及高致死致残的疾病[13],缺血性脑卒中治疗策略仍然不能满足众多患者,仅有的临床药物组织纤溶酶原激活剂药物因为狭窄的治疗窗口限制其临床普遍使用[11]。脑缺血神经系统紊乱的特征使研究者在开发新疗法时自然考虑到雌激素的神经作用。然而临床研究结果的矛盾性[14]以及雌激素对脑部已受损神经元作用微弱性[7]使目前药物在脑缺血临床试验未见其身影[13]。有趣的是作为雌激素新型受体,GPR30 保留雌激素的神经保护作用,并且相较于其他已知的选择性以及非选择性雌激素受体调控剂,GPR30 激动并不会造成依赖雌激素受体激活参与的乳腺癌发病[15]以及其对生殖系统[16]的影响较少。这些特征使GPR30靶点在神经系统疾病中的应用得到广泛关注。
本综述介绍GPR30 受体分布,功能以及分子机制,进一步总结近10年来人们在几种神经系统疾病层面获得有关GPR30神经作用新认识,包括通过调控非神经元功能维持病理状态下神经系统稳态,这些作用可能为GPR30 受体在治疗这些神经系统疾病提供新的治疗策略。
1 GPR30受体在脑部分布
免疫组织化学和原位杂交组织化学表明GPR30受体在鼠类大脑中广泛分布[17],其中具有GPR30 高表达特征的脑区包括前脑的新旧皮质以及海马结构,间脑的下丘脑,中脑黑质致密部,脑桥和小脑部分区域(表1)。人脑GPR30受体分布特征与这些类似[18]。

表1 GPR30受体在鼠类大脑中高表达对应的区域[17]Table 1 Brain regions with high expression of GPR30 receptor in mice[17]
关于细胞类型,GPER 在神经元以及多种胶质细胞中均有表达。电子显微镜结构结合抗体标记揭示不同脑区神经元的GPER细胞内超微结构定位。在海马区域,GPR30 受体在椎体神经元中表现出核周围广泛分布的特征[19-22],包括细胞膜、内质网、核周围胞质、树突棘以及其突触后膜、轴突以及其突触前膜。在中脑纹状体中,该受体在胆碱能神经元的树突以及轴突分布[23]而在多巴胺能神经元突触前膜分布稀少。在下丘脑中,GPR30 受体在黄体生成素释放激素(LH-releasing hormone,LHRH)神经元[24]以及下丘脑室旁核(paraventricular nucleus,PVN)催产素神经元以及促皮质素释放激素神经元分布[25]。在垂体中,GPR30 受体在促性腺激素细胞,催乳素细胞以及生长激素细胞中均有分布[26]。另外对于具有干细胞特征,分化程度较低的细胞往往表现出GPR30能够入核的特征,比如肿瘤细胞[27]以及神经干细胞[28](neuron stem cell,NSC)。根据上述证据,一种猜测是当神经元成熟度较低时[28-29],GPR30 受体在细胞核中分布。随着逐渐成熟,GPR30受体从细胞核向胞体再向神经元其他结构比如轴突,树突中再分布。
2 脑部GPR30受体介导的信号通路及其产生的生物效应
作为GPCR 家族成员,GPR30 受体介导细胞内信号通路的过程与已知GPCR介导的经典细胞内信号相似(图1A)。首先其定位在某种膜结构上,然后与特定配体结合,膜内偶联的G蛋白内部发生不同亚单位解离,这些亚单位(如Gα)随后与其他蛋白相互作用触发细胞内信号级联反应。在细胞膜或者核周内质网分布的GPR30 受体激活后能够激活c-Src 蛋白,进一步导致基质金属蛋白酶(matrix metalloproteinase,MMP)向细胞外转运(图1A)。MMP 随后在细胞外释放与细胞质基质结合的EGF,这些因子与细胞膜外侧受体比如EGFR结合调控下游不同MAPK亚族,P13K/Akt信号通路完成转激活事件。这些转激活事件产生的效应具有脑区细胞特异性。在海马神经元中,GPR30/MAPK 与GPR30/P13K/Akt 信号通路均能够参与调控记忆功能。GPR30/ERK在CA1 区域突触后膜上增强NMDA功能以及随后AMPA受体在突触后膜的定位,这些效应能够促进突触后膜对兴奋性神经递质的增敏性[30](图1N)。在海马腹侧区域,ERK信号的激活程度往往较弱[31],GPR30 受体调控记忆的分子机制主要与另一MAPK 亚族JNK 有关[32]。GPR30/P13K/Akt 信号通路也能够促进LTP 形成需要的AMPA 受体突触后膜定位[33-34],不过这种效应与促进突触后膜蛋白定位的骨架蛋白表达有关(图1M)。另外该信号通路还能通过调控actin 蛋白解聚行为调控树突密度[34](图1M)。值得注意的是得到GPR30/P13K/Akt 对海马区域效应特征的研究采用系统给药方式[34],这使GPR30与P13K/Akt 信号通路之间在海马中可能不是直接上下游关系。比如通过激动苔藓纤维-CA3突触区域的GPR30 受体发现该受体通过BDNF/TrKB与P13K/Akt信号通路建立联系[35]。在皮质神经元中,GPR30/ERK信号通路能够通过阻碍DAPK1 对NMDA 受体ser1303 位点的磷酸化过程增强皮质神经元对谷氨酸耐受性[22](图1B)。在下丘脑视上核以及室旁核中,GPR30激动反而抑制ERK信号,进一步造成一氧化氮合酶(nitric oxide synthase,NOS)功能抑制,该效应可能参与下丘脑对血压的调控过程[36](图1F)。
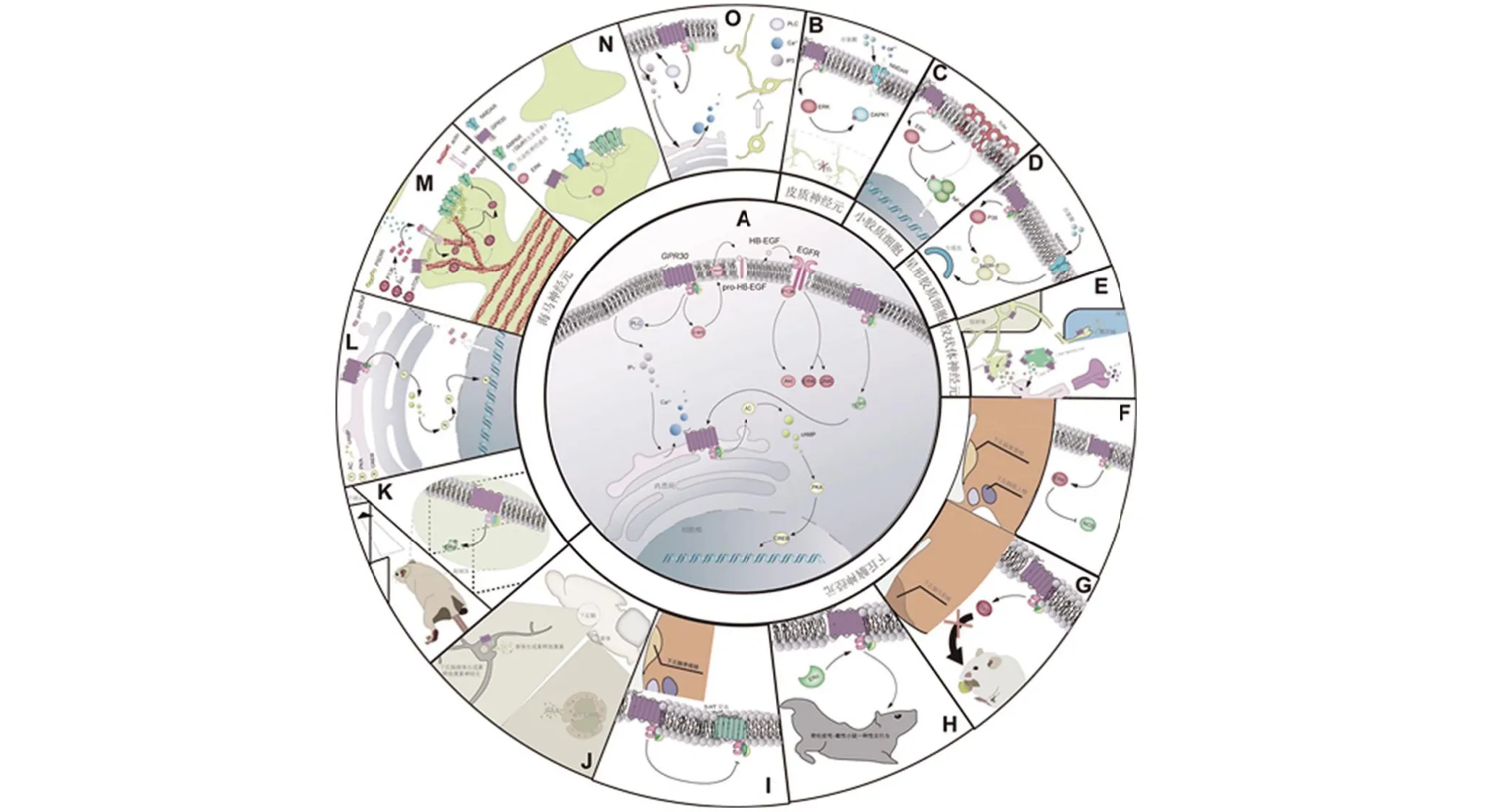
图1 脑部GPR30受体介导的信号通路及其产生的生物效应Figure 1 Signaling pathways mediated by brain GPR30 receptors and their biological effects
在其他神经细胞中,如小胶质细胞,GPR30/ERK抑制NF-κB 磷酸化进而抑制小胶质细胞炎性表型形成[37](图1C)。GPR30 受体的抗炎作用还能通过GPR30 调控TLR4 表达抑制NF-κB 信号通路实现[38](图1C)。在星形胶质细胞中,GPR30 激动调控的MAPK 亚族为P38,GPR30 通过抑制P38 信号恢复接受谷氨酸刺激的星形胶质细胞自噬水平[39](图1D)。
除了依赖转激活的信号通路,细胞膜以及内质网上的GPR30受体还能通过偶联的Gs蛋白亚基直接引发PLC/IP3 以及AC/cAMP/PKA 信号通路。GPR30/IP3信号通路被证实能够显著增加小鼠胚胎18天海马区域神经元内的钙信号以及神经突长度[29](图1 O)。GPR30/AC/cAMP/PKA信号通路在海马脑区激活后能够进一步激活下游转录因子CREB改善去卵巢伴随铁损伤的雌鼠物体识别以及情绪记忆能力[40](图1L)。在成年雄性大鼠海马苔藓纤维-CA3 突触中,GPR30/PKA 能够促进CA3 区域的BDNF 分泌以快速诱导长时程抑制的形成[35](图1L)。CA3区增加的BDNF通过Trkb信号通路激活Akt/mTOR使突触外的AMPA受体数目减少从而达到短期沉默突触的效应[35](图1M)。
在下丘脑弓形核神经元中,GPR30 激动后可以通过非转激活作用快速激活STAT3,随后激活的STAT3启动基因组效应参与诱导雄鼠厌食行为形成[41](图1G)。此外,该区域的GPR30在雌鼠中还参与调控弓形核-内侧视前核轴功能以促进雌性大鼠性交行为[42]。其他不清楚信号通路特征的GPR30 功能还包括正向调控下丘脑-垂体轴释放激素过程,比如促进下丘脑LHRH神经元释放促黄体生成素释放激素[24],垂体催乳素细胞释放催乳素过程[26](图1J);使PVN 区域对5-HT 脱敏[25,43]并造成下游靶组织垂体分泌更少的催产素以及促肾上腺皮质激素[25](图1I);GPR30的5-HT受体信号抑制作用有望增强选择性血清素再摄取抑制剂抗抑郁作用[25];促进纹状体区域胆碱能神经元向海马输入乙酰胆碱[44](图1E);直接调控纹状体中间GABA能神经元或者胆碱能神经元与多巴胺能神经元之间的相互作用,间接调控多巴胺能神经元释放多巴胺[23](图1E)。
另外脑部其他雌激素受体被发现与GPR30 受体互作,并且这些作用具有脑区以及性别特异性。在雄鼠海马垂直区域中,GPR30受体激活能够促进雄性小鼠该区域的ERα磷酸化[45],并且该效应参与减少雄鼠在高架十字迷宫中焦虑行为(图1K)。而在雌鼠下丘脑腹内侧,GPR30受体作为ERα的下游参与调控鼠性交行为[46](图1H)。除了串联的作用方式,GPR30 与ERα还能以并联的方式共同调控bcl2 的表达[18]。总之,GPR30受体作为一种GPCR,其保留GPCR经典信号通路途径,包括转激活以及非转激活途径,这些信号通路在大脑不同细胞中分别介导神经可塑性,神经内分泌稳态,心血管自主神经调节,能量摄取以及神经炎症等生物效应。
3 GPR30受体在神经退行性疾病中的作用
3.1 阿尔兹海默病 痴呆被定义为一种由多种疾病参与造成的临床综合征,该综合征临床上能够表现出一种乃至多种认知功能障碍,如严重失语,记忆丢失[47]。具有记忆丢失特征的痴呆患者在过去临床上被定义为阿尔兹海默病(Alzheimer's disease,AD)[47]。另外阿尔兹海默病还可以通过其病理去定义:尸检确定具有脑部中细胞外有Aβ沉积导致的淀粉样沉积以及神经元内tau 构成的神经纤维缠结。然而随着研究不断深入,AD的定义被限定在病理方面而排除临床[48]:在活体中被同时诊断出具有Aβ以及病理tau生物标记物[49]。这些生物标记物在个体中随着多种风险因素的介入,比如年龄,性别,它们一起形成一种复杂的、渐进的且多种脑内细胞功能障碍和死亡的过程[50],最终推动个体从临床前进入临床阶段,演变为痴呆[51]。目前临床仅支持对症治疗,包括使用乙酰胆碱酯酶抑制剂和NMDA 受体抑制剂通过矫正脑部神经递质释放紊乱在一定程度上缓解认知的衰退[12]。然而AD神经病理不能被这些药物所改善,随着时间推移,它们会再次联合诸多因素导致认知不可逆的恶化。这一局限迫使目前研究集中开发修饰疾病的疗法,这些疗法可以阻止AD 这一生物条件的恶化,还有可能具有对症治疗的疗效[12]。目前诸多文献报道激动GPR30受体对动物具有增强认知功能的作用。在早期一项延迟位置匹配T 迷宫(delayed matching-to-position T-maze,DMP T-maze)实验中,接受GPR30激动剂G1腹腔注射的去卵巢大鼠的学习能力显著增强,表现为完成任务规定所需要的时间周期显著缩短[52]。这种学习加速的过程与记忆功能的变化相关。记忆主要包括陈述性记忆,工作记忆以及非陈述性记忆[53],研究报道G1在去卵巢鼠中影响的记忆类型主要是陈述性记忆。在Y 迷宫实验中,G1能够显著增加去卵巢大鼠在延迟期的空间识别记忆[54]。在水迷宫实验中,G1同样能够改善空间识别记忆,使去卵巢小鼠在目标象限区域的停留时间恢复至与空白组近似,并且小鼠记忆的变化与小鼠学习过程变化一致,后者表现出逃避潜伏期显著缩短的特征[34]。AD模型参与的研究为调控GPR30作为潜在的AD 对症治疗策略直接提供有力的支持。G1 对陈述性记忆的改善作用在非卵巢摘除5XFAD小鼠复现,表现为物体识别记忆功能受损得到一定程度缓解。并且这种认知改善作用具有性别差异,表现为雌鼠易感,而雄鼠无响应[55]。在机制方面,G1 激动对空间记忆学习的影响至少涉及两方面:(1)直接调控基底前脑胆碱能系统间接影响海马脑区功能。免疫组化结果表示GPR30在去卵巢SD大鼠的基底前脑胆碱能神经元中表达,给予G1能够增加海马区域乙酰胆碱含量,伴随认知功能的改变,而这些作用在使用GPR30拮抗剂G15 后减弱[56]。(2)调控海马区域神经元突触可塑性。分子生物学研究表示在海马中表达的GPR30 被激活后能够通过SRC-1/P13K/mTORC2/Akt信号通路促进与突触相关蛋白表达,比如位于突触前膜蛋白Spino,突触后膜蛋白PSD95 和谷氨酸受体GluR1,其中PSD95 的表达有利于GluR1 突触后膜锚定[33-34]。另外该信号通路还能够影响突触中的actin 骨架蛋白聚合以及解聚行为进而调控树突棘密度[34]。类似地,脑室注射G1同样能够通过调控海马CA1区的actin骨架蛋白聚合改善记忆[57]。不过由于给药方式的不同,该研究表示造成该效应的分子机制与JNK 信号通路激活有关,而非ERK以及P13K/Akt信号通路[32]。最近一项细胞研究表示GPR30激动对AD病理下神经元具有一定保护作用[58]。G1能够改善Aβ1-42刺激下的大鼠皮质神经元活力。这种挽救行为可能与GPR30 激动降低胞内氧化应激水平有关[58]。上述研究表明GPR30对神经元功能具有保护和增强作用,这些作用可能将来被用于研发AD对症治疗策略。在AD中,非神经元介导的病理事件同样影响认知功能。尽管目前没有直接研究表示GPR30能够调控AD脑内固有免疫异常响应,在其他疾病研究中,GPR30 激动具有显著的中枢抗炎作用[38]。同时GPR30 还能够影响星形胶质细胞对神经元作用[59]。这些证据提示GPR30靶点的开发可能是AD疾病修饰策略的新方向。
3.2 帕金森病 PD是继AD最常见的神经紊乱,其病理特征主要表现为在黑质致密部出现多巴胺能神经元丢失,这种紊乱会进一步造成该脑区投射的纹状体区域多巴胺含量锐减,最终导致高级运动功能发生障碍症状出现[60]。国内老龄化带来的一个问题是PD发病率的提高,2020年底中国预计将达到362万左右群体患上PD,占全中国60 岁以上老年人群体的1.37%[61]。与AD 不一样的是,PD 是可治疗的,PD 的对症治疗策略治疗效率要比治疗AD的对症治疗好很多[62]。目前的临床对症治疗策略包括改善运动症状以及改善早期非运动症状的策略,而改善运动症状的策略又可以根据患者对一线用药左旋多巴的耐受程度进行分类[63]。目前的临床前研究提示GPR30 的神经保护作用能有效保护黑质纹状体多巴胺能通路,同时其还能有望用于开发治疗改善PD 自主神经功能障碍,尤其是消化系统症状,比如便秘。Bourque 等[16]发现G1 药理激动GPR30 能够改善MPTP 中度损伤导致的纹状体区域DA代谢周期运转上调以及多巴胺能神经元末端丢失。该研究中选用的MPTP诱导模型[16]除了具有中枢样退变外还具有肠道神经丢失和外周固有免疫异常激活的病理特征[64],这些外周异常在处于早期PD的患者中出现并且它们参与介导该群体胃肠道运动障碍症状的形成(比如便秘)。该研究团队表示激动GPR30 带来的多巴胺能神经保护作用对该模型的肠道内肠肌丛中的多巴胺能神经元同样有效[64]。另外,腹膜腔巨噬细胞以及外周循环单核细胞也是GPR30 的重要调控对象。Iba1 免疫组化以及体视学结果表明G1能够减少腹膜腔上的巨噬细胞数目。而体外内流式细胞术实验结果表示G1 给予能够直接缓解血液中单核细胞群体表型向促炎方向转换。这些结果提示激动GPR30还可能应用于治疗PD早期阶段出现的肠道症状[64]。临床研究表明PD 除了具有异常中枢外周炎症外,还存在神经营养因子中枢含量减少的特征[65],该特征也与PD经典病理黑质纹状体通路多巴胺能神经元衰退有关。体内药理研究表明GPR30激动能够增加MPTP 模型脑BDNF 含量并抑制GSK-3β活性,从而发挥神经保护作用[66]。
4 GPR30受体在耐药性癫痫中的作用
癫痫被定义为脑局部或者整体存在异常同步的神经元活动[67-68],并且这些活动能够引发多次临床可观察的发作[69]。目前抗癫痫发作类药物(Antiseizure Drug)依旧是治疗癫痫的首选药物,然而目前存在近30%的患者对该类型耐受[67]。抗癫痫发作类药物耐受的机制主要包括靶点假说,转运体假说以及神经元网络假说[70]。其中神经元网络假说为GPR30 开发为治疗癫痫的新靶点提供参考。该假说认为耐药癫痫患者脑部不只是神经元放电异常,其还伴随胶质增生,神经退行性发生以及轴突延伸障碍导致的神经环路异常[70]。几项研究表示GPR30 的神经保护作用还可以用来改善耐药性癫痫当中的神经退行性发生以及神经炎症。
Kurt 等[71]发现GPR30 激动具有促进癫痫发生的作用。研究者在PTZ 诱导的点火模型中发现雌激素与GPR301激动剂能够显著增强该模型的发作。进一步的生化分析表现出G1 给予显著促进海马以及皮质区域NO水平表达,提示G1可能通过改变脑部氧化应激过程诱导神经炎症。随后Zuo 等[72]得到相反的结果,其认为可能与研究选择的不同癫痫诱导模型以及G1 浓度有关。该项研究通过毛果芸香碱构建耐药性癫痫模型并通过敲除GPR30 排除了药效学研究带来的药物浓度变量对实验结果的干扰[72]。GPR30敲除显著增强小鼠发作程度以及缩短痉挛潜伏期。病理分析结果提示该模型中出现海马区神经元损伤和神经炎症。这些结果初步表示GPR30 的功能失调可能参与具有海马硬化特征的耐药性癫痫患者发作。相同模型的药理干预结果提示GPR30 激动能够改善耐药性癫痫中的认识功能[73]。给予未敲除GPR30 的小鼠G1 后研究团队发现G1 能够有效改善小鼠认知功能,并伴随有效控制海马苔藓纤维发芽。最近的临床研究建立了GPR30 与皮质区功能异常相关的癫痫发生的关系[74]。首先Wang 等[74]观察到该类癫痫患者脑部神经元以及小胶质细胞的GPR30 表达下调。就神经元方面,研究者进一步通过细胞实验揭示了GPR30激动能够增强原代皮层神经元PKA以及磷酸化PKA的表达,另外GPR30 还能够通过改变NR2A/B 表达调控原代皮质神经元突触后兴奋性电流自发频率。而小胶质细胞方面,小胶质细胞GPR30表达下调与NF-κB信号激活有关。另外该研究发现采用非侵入性手段PET-CT观测女性患者脑内葡萄糖能量代谢特征能够有效推测GPR30 表达情况,该策略有望进一步将GPR30 开发成能够反映皮质发育不全患者特定疾病进展的新分子标记物[74]。总之,海马皮质区域GPR30表达的下调可能促进耐药性癫痫病理发生发展[72-74],而通过适当激动GPR30 可能能够有效增加脑部GPR30表达水平从而改善疾病进展[73]。
5 GPR30受体在脑缺血中的作用
脑缺血是一类因脑部供血不足造成神经元功能障碍甚至死亡的的神经系统紊乱[75]。脑部血流量的降低原因包括斑块介导的栓塞或者循环系统功能障碍[76]。其中前者引起的脑缺血为局部脑缺血,又称为脑卒中。由心脏功能异常造成的脑缺血则为全局脑缺血[77]。目前临床上分别采用溶栓剂以及抗氧化或神经炎症药物分别治疗脑缺血中的早期急性缺血事件以及缺血后事件[75]。尽管理论上雌激素的神经保护作用本身对缺血后事件可能有益,然而一项早期临床研究表示绝经后女性脑卒中对雌激素替代疗法收效甚微,甚至该策略加重该群体致死率[14]。有趣的是作为雌激素受体,GPR30激动剂在最近关于治疗具有绝经特征的脑缺血动物模型的报道中表现出优异的效果。
在人为绝经雌鼠中MCAO 损伤诱导后短时间内单次给予G1 抑制神经元缺血性损伤事件的发展[38]。进一步研究表示该作用可能与降低小胶质细胞炎性激活信号通路TLR4-NF-κB 有关[38]。与小胶质细胞类似,星形胶质细胞的GPR30在局部脑缺血环境下也具有神经保护作用[39]。在该细胞背景下激活能够抑制炎症响应并且恢复自噬水平并抑制反应性星形胶质细胞增生[39]。
在绝经期动物模型合并全局脑缺血病理中,海马区域不同细胞中的GPR30 均表达下调[78]。与局部脑缺血类似,全局脑缺血中的小胶质细胞的GPR30表达下调能够增强该类细胞的炎性活动[20,79],这种活动能够被G1 激动抑制。而在神经元中,GPR30 被发现能够允许健康神经元表达更多IL1β受体内源性拮抗蛋白IL1RA 以增强其对神经炎症耐受性[20]。当神经元GPR30 表达下调时,神经元的对小胶质细胞来源的IL1β耐受性下降,进而引起神经元死亡并释放DAMP,这些抗原被小胶质细胞的TLR4 识别后激活,促进更多的促炎因子释放,形成恶性循环,加重梗死区域的扩散[20]。相同病理背景下的星形胶质细胞中,其GPR30 的激活能够促进非炎性表型反应性星形胶质细胞形成[59],这些细胞能够增强自身芳香合成酶-脑源性雌激素信号通路合成并释放脑源性雌激素促进海马区齿状回以及CA1 区域的NSC 增殖并发育成成熟神经元。
总之,脑部多种细胞的GPR30在绝经期不同程度脑缺血中表达或功能下调,而恢复或增强其功能能够带来神经炎症抑制,健康神经元耐受性增强以及神经元再生等效应,这些效应有利于改善疾病进程。
6 讨论
GPR30 受体在中枢系统中广泛表达以及其能够调控多种初始发病机制不同的神经系统疾病共同的下游病理事件的特征提示该靶点的神经保护作用可能具有泛神经系统疾病改善的特征。根据这一猜想,最近有综述推导出GPR30 激动还可能用于治疗亨廷顿疾病以及多巴胺能拮抗药物治疗精神分裂引起的迟发性运动障碍[60]。然而目前靶向GPR30 对多种神经系统疾病独特的病理特征的影响依旧无文献支持。在PD领域中,现有的药理学证据均来自MPTP模型,该模型能够破坏神经元呼吸链正常运作,进一步造成线粒体氧化应激,最终能够在较短的时间内造成多巴胺能神经元大量丢失[80]。然而这种模型不能模拟PD另一经典病理特征—α-突触核蛋白堆积形成lewy小体[63],这提示目前采用MPTP 模型获得的药效结果可能在临床转化上受到限制,仅对具有神经炎症或者线粒体氧化应激明显的PD亚型患者可能有效[81],而对那些具有α-突触核蛋白基因突变的患者的效应还需要在具有lewy 小体病理特征的转基因动物上进一步确定。同理,对于具有AD 病理特征的痴呆患者以及亨廷顿病患者来说,观察GPR30 对Aβ或者tau病理以及亨廷顿蛋白堆积改善是十分有必要的[82]。除此之外,前述证据指出靶向GPR30受体更可能作为一种临床辅助策略,比如在治疗耐药性癫痫以及PD 早期症状的时候,GPR30 激动剂可能可以联合现有标准疗法治疗而不是替代这些策略。
尽管目前GPR30 受体被普遍认为是一种雌激素受体,然而其激动产生的独特的机制以及随后表现出包含雌激素神经保护作用同时排除直接给予雌激素带来的副作用的效应引发了关于其是否是雌激素受体的讨论[2]。除此之外,GPR30效应的工具药的靶点非特异性以及生理环境下激动GPR30所需的雌激素浓度过高等证据对该观点同样提出质疑[2]。对GPR30受体与雌激素受体的关系的进一步认识有利于将GPR30开发成合适的药物,以克服雌激素带来的多种副作用,如雌化效应、乳腺癌诱发、癫痫诱发以及脑卒中等。
猜你喜欢
作文周刊·小学二年级版(2022年20期)2022-05-05 01:33:06
神经损伤与功能重建(2020年11期)2020-12-01 05:01:54
创新作文(小学版)(2019年10期)2019-09-25 08:12:28
小学生学习指导(低年级)(2017年5期)2017-05-04 04:14:38
湖南中医药大学学报(2016年1期)2016-12-01 04:08:21
中国康复理论与实践(2015年10期)2015-12-24 05:42:43
磁共振成像(2015年1期)2015-12-23 08:52:21
吉林大学学报(医学版)(2015年5期)2015-12-16 15:43:56
中国体外循环杂志(2015年3期)2015-12-08 05:13:01
作文与考试·小学高年级版(2015年17期)2015-05-30 10:48:04
