
青檀扦插苗对不同氮素水平的形态、光合生理响应和转录组分析
2023-12-04 00:46孙忠奎李国华
南京林业大学学报(自然科学版) 2023年5期
郭 伟,韩 秀,张 利,王 迎,杜 辉,燕 语,孙忠奎,张 林,李国华*,罗 磊*
(1. 泰安市泰山林业科学研究院,山东 泰安 271000;2. 菏泽学院农业与生物工程学院,山东 菏泽 274015;3. 泰安时代园林科技开发有限公司,山东 泰安 271000)
氮素是植物生长发育所必需的大量营养元素,是氨基酸、蛋白质、叶绿素、植物激素和核酸等生物分子的基本元素之一[1-2]。植物可以吸收有机氮和无机氮,在土壤中植物根系主要通过硝酸根转运蛋白(nitrate transporter, NRT)和铵根转运蛋白(ammonium transporter, AMT)分别来吸收硝态氮(硝酸根)和铵态氮(铵根)为主要形式的无机氮[3],然后在根系中被同化利用,或者储存在根细胞的液泡中,或者运输到地上部叶片中被同化利用。
土壤中的氮素对植物生长和形态结构建成有显著影响,是限制植物生长的一个重要因素,植物在生长过程中会对氮素水平做出相应的形态和生理响应。对模式植物拟南芥(Aradidopsisspp.)的研究发现,随着氮素水平的降低,地上部生长变弱,根系生长加强,氮、总蛋白、氨基酸、可溶性糖以及淀粉含量均降低[4]。对木本模式植物杨树的研究发现,缺氮处理能够促进杨树根系生长,抑制地上部生长和光合作用,减少根和叶片的氮和氨基酸含量,而高氮处理抑制根系生长,促进地上部生长和光合作用,增加根和叶片的氮和氨基酸含量[5-6]。
植物对环境变化做出的形态和生理响应受到基因的调控,而进行转录组测序是研究基因表达模式的高效手段。分析拟南芥响应不同氮素水平的转录组结果发现,超过2 000个基因在不同氮素水平响应中发生表达变化,整合基因调控网络分析揭示了硝酸盐响应基因和11个高共表达基因簇之间的关系,其中4个基因簇在硝酸盐响应过程的转运、信号转导、代谢中起到重要作用,bZIP和G2-like转录因子是硝酸盐转运和信号转导中的重要调控因子[4,7-8]。分析杨树响应不同氮素水平的转录组结果发现,在缺氮和高氮处理条件下,1 064个基因差异表达,离子转运、生长素刺激物、脱落酸刺激物相关的基因本体得到富集,基因共表达网络分析分别鉴定得到缺氮和高氮处理的58和51个中枢基因(hubgene),推测杨树通过启动胁迫响应通路来对生长发育进行相应的调控[5]。
青檀(Pteroceltistatarinowii)是我国特有的榆科青檀属的单种属落叶阔叶乔木[9],在石灰岩山地的生态修复中起到了显著的水土保持作用[10]。氮素对树木生长和形态结构建成有显著影响,是陆地环境中林木生产力的制约因素[11]。但是,作为耐瘠薄的水土保持先锋树种,青檀在氮素响应条件下的生长发育分子机制研究至今鲜见报道。本研究以青檀为研究材料,进行不同浓度氮素处理,对形态发育和光合生理指标进行分析,并利用3代和2代高通量转录组测序技术,筛选氮素响应相关基因,为深入开展青檀基因功能的研究提供参考。
1 材料与方法
1.1 植物材料、生长条件和处理方法
试验地设在山东省泰安市泰安林业协同创新管理中心植物培养室,光照强度350 μmol/(m2·s),光周期16 h光照/8 h黑暗,温度25 ℃,湿度40%~50%。青檀供试材料来自同一母株,由泰安市乡土观赏树种国家林木种质资源库(36°11′12″N,117°09′5″E)提供。2019年5月27日,在间歇喷雾条件下,选取18~22 cm长的插穗在珍珠岩和蛭石体积比1∶1的基质中进行扦插,2019年6月24日,对生根扦插植株进行为期4周的5 mmol/L NH4NO3的Hocking’s营养液处理(每周2次,周二和周五9:15进行),2019年7月22日,随机选取63株高为40 cm的扦插生根植株,用蒸馏水将根洗干净后转移至装有基质的花盆中,在间歇喷雾条件下进行缓苗。完成缓苗后,在2019年7月29日,将63株青檀植株随机分为3组,每组21株,每3株放在一个塑料托盘上,进行3组施氮处理试验:缺氮(N0)、中氮(N2)、高氮(N50),分别施用含有0、2、50 mmol/L NH4NO3的Hocking’s营养液(每周2次,周二和周五9:15进行),每次将2 L营养液倒进塑料托盘,静置1 h,然后将剩余的没有被吸收的营养液导出。首次分组进行施氮处理前,每个花盆内插入1根与青檀植株等高的竹签,用来判断施氮处理前后的株高。首次进行分组施氮处理的日期设为第0天。
1.2 指标测定
第28天,从每个处理组的21株植株中随机选取6株植株,根据叶间隔指数(leaf plastochron index,LPI, 公式中记为ILPI)选取3片成熟叶片(ILPI为5~7, LPI0的叶片长度设定为小于2 cm),用Li-6400光合作用系统(LI-COR,Inc,Lincoln,NE,美国)测定净光合速率、气孔导度和蒸腾速率,用乙醇提取法测定叶绿素和类胡萝卜素含量。
从每个处理组随机选取6株植株,进行生长量、生物量和根系形态指标测量。株高使用直尺测量,地径使用游标卡尺测量。根系用清水冲洗两次后用吸水纸蘸干,用WinRHIZO根系分析系统测量总根长、总根体积、总根表面积。选取3片成熟叶片(ILPI为5~7),用Li-3100A叶面积仪(LI-COR, Lincoln, NE, 美国)测量叶面积,计算比叶面积(specific leaf area, SLA)。用电子天平测量地上部分(叶和茎)和地下部分(根)的鲜质量,然后在105 ℃杀青30 min,在85 ℃烘72 h至恒质量,测量干质量。总生物量为地上部分和地下部分生物量之和。根冠比为根系与地上部分的干质量之比。比根长为根长与根生物量之比。
从每个处理组随机选取6株植株,采用硫酸-过氧化氢消解,半微量凯氏定氮法分别测定根、叶片、茎的全氮含量。
利用SPSS 22.0的单因素方差分析(One-way anova)对不同处理组的形态发育和光合生理指标进行统计分析,采用Turkey法比较不同处理组间的差异性。
1.3 样本收集、RNA提取
在第28天对每个处理组的3株(生物学重复)青檀植株的根分别进行取样,放入锡箔纸包裹,液氮速冻后放入超低温冰箱保存。用RNeasy Plant Mini kit试剂盒(Qiagen,Valencia,CA,美国)进行RNA提取,用RNase-free DNase I(Qiagen,Valencia,CA,美国)去除RNA中的基因组DNA污染,然后用RNA MinElute kit试剂盒(Qiagen,Valencia,Carlsbad,CA,美国进行纯化)。用Qubit©RNA Assay Kit in Qubit©2.0 Flurometer(Life Technologies,CA,美国)检测RNA浓度,用RNA Nano 6000 Assay Kit试剂盒(Agilent Technologies,Santa Clara,CA,美国)通过Agilent 2100 Bioanalyzer系统(Agilent Technologies,Santa Clara,CA,美国)检测RNA的完整性。
1.4 PacBio文库构建、测序和数据处理
将3个处理组共9株青檀植株根的RNA进行等量混合,取5 μg混合RNA用于3代测序PacBio文库构建。分别使用SMARTer PCR cDNA Synthesis Kit试剂盒 (Clontech,Palo Alto,CA,美国)和Advantage 2 PCR Kit试剂盒(Clontech,Palo Alto,CA,美国)合成cDNA的第1和第2条链。通过BluePippin DNA Size Selection System(Sage Science,Beverly,MA,美国)生成并选择大于4 kb和小于4 kb的cDNA片段,利用SMRTbell Express Template Prep Kit 2.0试剂盒(Pacific Biosciences,Menlo Park,CA,美国)进行文库构建,通过Sequel System(Pacific Biosciences,Menlo Park,CA,美国)进行测序反应。
利用SMRTlink 7.0软件,设置参数“min_length 50,max_drop_fraction 0.8,no_polish TRUE,min_zscore 9 999.0,min_passes 1,min_predicted_accuracy 0.8,max_length 15 000” 进行数据处理,得到子读序(subread)和环形一致性序列(circular consensus sequencing read,CCS)。根据序列末端是否包含5′引物、3′引物或poly A序列来判断序列是否为全长序列。将同一转录本的全长非嵌合序列(full length non-chimeric read,FLNC)通过hierarchical n*log(n)算法聚类,得到一致性序列(consensus sequence,CS)。利用Arrow软件对一致序列进行校正,设置参数“hq_quiver_min_accuracy 0.99,bin_by_primer false,bin_size_kb 1,qv_trim_5p 100,qv_trim_3p 30”,得到校正一致性序列(polished consensus sequence,PCS)。利用二代转录组测序数据通过LoRDEC软件对校正一致性序列再次进行校正[12]。最后,利用CD-HIT软件[13],设置参数“-c 0.95-T 6-G 0-aL 0.00-aS 0.99”移除冗余序列,得到基因序列。
通过机器学习的算法,用相关物种的蛋白序列对ANGLE pipeline进行训练,使其了解密码子使用频率和蛋白质结构。用ANGLE pipeline寻找转录本的蛋白质编码区(CDS),通过是否含有起始密码子和终止密码子来判断CDS是否编码完整蛋白序列[14]。
为了对基因功能进行注释,利用Diamond(e-value阈值1e-5)比对NR(NCBI non-redundant protein sequences)、NT(NCBI non-redundant nucleotide sequences)、KOG/COG(clusters of orthologous groups of proteins)、KEGG(Kyoto Encyclopedia of genes and genomes ortholog)数据库[15-16],利用Hmmscan软件比对Pfam(Protein family, http://pfam.xfam.org/)数据库[17],利用Pfam数据库的比对结果,比对GO(Gene Ontology)数据库,获得相应的注释信息[18]。
1.5 cDNA文库构建、测序和数据处理
3个处理组共9株青檀植株的根的RNA分别用TruSeq RNA sample preparation kit试剂盒(Illumina, San Diego, CA, USA)进行2代高通量测序cDNA文库的建立。首先用Oligo-(dT)磁珠从5 μg总RNA中纯化得到mRNA,利用Elute-Prime-Fragment Mix 裂解液将mRNA打成120~200 bp的短片段。以mRNA短片段为模板,利用随机引物反转录合成单链cDNA,然后利用DNA聚合酶I合成双链cDNA,修复末端并连接接头后,通过PCR反应扩增,产物经过纯化后,用Illumina HiSeqTM 4000(Illumina, San Diego, CA, USA)进行测序反应。
用Bowtie2软件将过滤掉测序数据中低质量、污染接头后的有效读序(clean read)与3代测序得到的转录本相比对,用RSEM软件统计比对结果,计算每百万测序碱基中每千个转录本测序碱基包含的片段数量(FPKM)来得到基因的表达水平。利用R语言的DESeq2软件包,根据阈值False discovery rate(FDR)< 0.05 和|log2(表达量差异倍数)|>1筛选差异表达基因(differentially expressed gene)。利用KOBAS 3.0对差异表达基因进行KEGG通路富集分析。
1.6 相对荧光定量PCR表达分析
以800 ng RNA作为反转录模板,用Invitrogen的Superscript Ⅲ First-Strand Synthesis System反转录试剂盒(Invitrogen,San Diego,CA,美国)合成cDNA,稀释20倍后用于荧光定量 PCR反应。使用荧光定量试剂盒TB Green©Premix Ex TaqTM(TaKaRa,大连,中国),根据试剂盒要求设计引物见表1。

表1 荧光定量PCR的引物序列
通过ABI 7500实时定量PCR系统(Applied Biosystems,Carlsbad,CA,美国),进行相对荧光定量PCR实验。每个试验设3次重复,选取转录起始因子(transcription initiation factor,Pt12965/f6p0/1249)作为内参基因。利用Sequence Detection Software(2-ΔΔCt法)[19]进行数据分析。
2 结果与分析
2.1 不同氮素水平对青檀生长的影响
氮素水平的提高有利于青檀植株地上部分生长,呈现出青檀植株的株高、地径、叶片数量、叶面积、比叶面积、叶片生物量、茎生物量随着氮素浓度提高而提高的趋势,而根系的生长却恰恰相反,总根长、总根表面积、总根体积、比根长、根冠比、根系生物量随着氮素浓度提高而降低。同时,高氮素水平处理明显促进了叶绿素a、叶绿素b、类胡萝卜素含量的提高,增强了净光合速率和气孔导度。蒸腾速率在缺氮处理时显著降低,却在高氮处理时变化不显著。植株根系、叶片、茎的氮含量的变化趋势与氮素水平高低一致,高氮素水平处理时氮含量最高,缺氮处理时最低(表2)。
2.2 3代全长转录组测序以及功能注释
3代全长转录组测序共产生23 007 808 条子读序(subread),平均长度为1 098 bp,N50值为1 362 bp。通过软件进行数据处理后,分别产生313 170条环形一致性序列(CCS)、219 220条全长非嵌合序列(FLNC)、29 644条校正一致性序列(PCS)(表3)。利用2代转录组测序数据进行校正后,最终得到11 801 条基因,平均长度为1 375 bp,N50值为1 628 bp。将11 801条基因序列在7个数据库中进行比对,结果显示分别有11 052(93.65%)、10 261(86.95%)、7 534(63.84%)、6 895(58.43%)、9 280(78.64%)、10 990(93.13%)、7 534(63.84%)条基因在NR、NT、Pfam、KOG、Swiss-Prot、KEGG、GO数据库中得到基因功能注释,11 232(95.18%)条基因至少在一个数据库中得到注释,5 054(42.83%)条基因在全部7个数据库中得到注释。
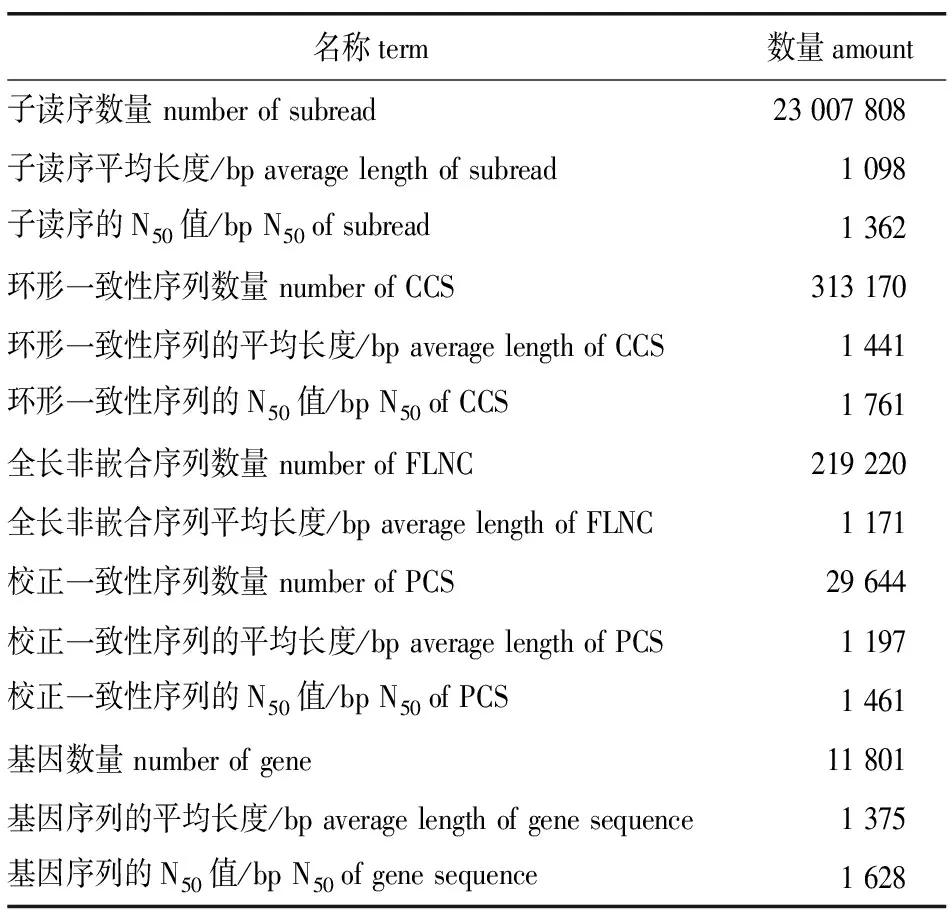
表3 青檀3代测序数据统计
2.3 不同氮素水平处理青檀植株的转录组分析
原始数据经过过滤,共生成了538 947 568条有效读序(clean read),其中,302 754 292(56.18%)条能够与青檀3代测序转录组数据库匹配(表4)。
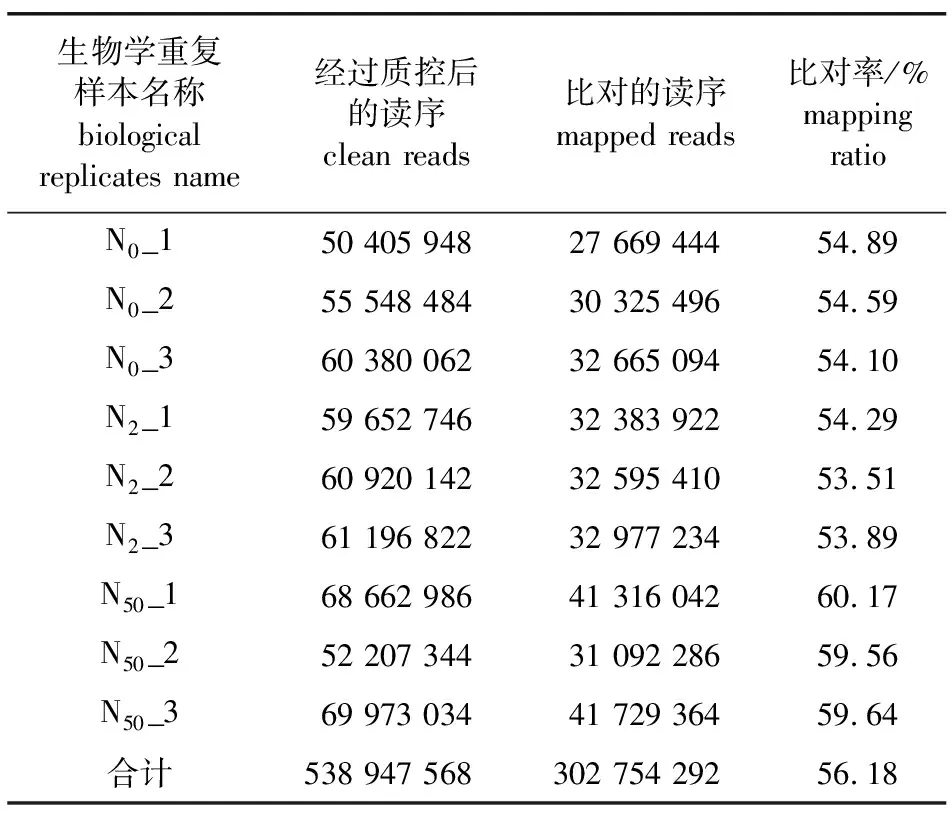
表4 不同氮素处理2代转录组测序结果统计
对N0、N2、N50的表达水平分布进行统计显示,发现大部分数值出现在1.2~1.9处,中位数出现在1.5~1.6处(图1)。对不同处理、不同生物学重复的表达量进行Pearson相关性检验,结果显示相同氮素水平处理的不同生物学重复之间的相关性较高,相关系数均接近1,最小值为0.961,说明相同氮素水平处理的不同生物学重复的表达模式非常相似;不同氮素水平处理间样本的相关系数明显低于同一氮素水平处理间样本的相关系数,证实了试验的可靠性和样本选择的合理性。

N0、N2和N50分别代表缺氮、中氮和高氮。下同。基因表达量用log10(FPKM+1)进行标准化处理。N0、N2 and N50 represent limiting, intermediate and luxuriant nitrogen levels. The same below. Gene expression levels is normalized by log10(FPKM+1) algorithm. 图1 不同氮素水平处理的相关性分析及表达量统计Fig. 1 Correlation analysis and expression statistics for treatments at different levels of nitrogen
对不同处理之间样本的转录组数据进行比对分析,如图2所示,氮素水平差异越大,产生的差异表达基因的数量就越多,N50与N0比对有最多的差异表达基因的数量1 851个,其次是N50与N2比对有1 613个差异表达基因,最后是N2与N0比对有1 525个差异表达基因。N50与N2和N2与N0比上调差异表达基因集合的交集是76个基因,它们都属于N50与N0比上调差异表达基因集合,说明这76个基因随着氮素水平提高稳定上调表达;N50与N2和N2与N0下调差异表达基因集合的交集是32个基因,它们都属于N50与N0比下调差异表达基因集合,说明这32个基因随着氮素水平提高稳定下调表达;不存在属于N50与N2和N2与N0比上调或下调差异表达基因集合的交集却特异于N50与N0比上调或下调差异表达基因集合的基因。这108个基因在不同氮素水平处理的比对中均存在表达差异,暗示其在不同氮素水平均存在重要的调控作用。
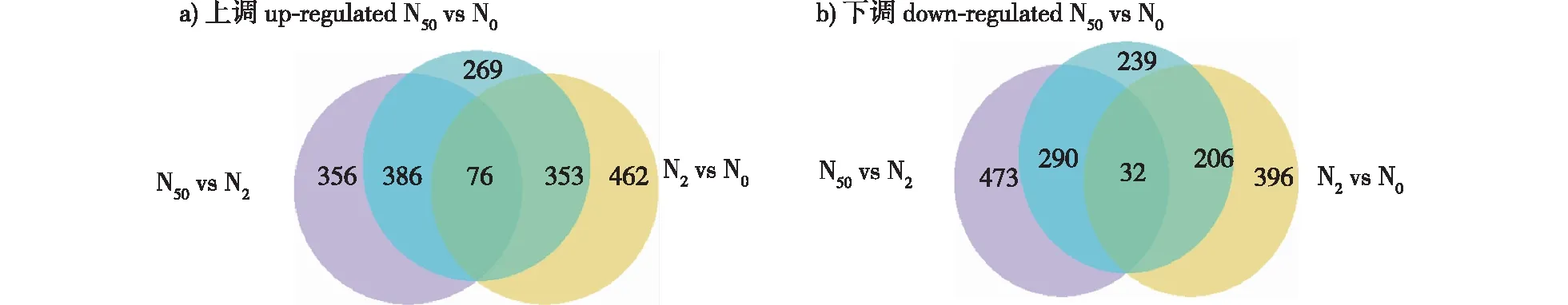
图2 不同氮素水平处理比对上调和下调差异表达基因韦恩图Fig. 2 Venn diagrams presenting differentially expressed genes up-regulated and down-regulated in the comparisons of treatments at different levels of nitrogen
2.4 不同氮素水平处理表达差异基因的KEGG通路富集分析、表达分析
对在不同氮素水平处理的比对中均存在表达差异的108个基因进行KEGG通路富集分析。可见基因富集于35个代谢通路。对排名前20的代谢通路进行分析结果(图3)表明,排名前两位的分别是光合作用(photosynthesis)和氮代谢(nitrogen metabolism),富集因子分别是0.095和0.100,富集的基因数量分别是6和3。基因注释表明光合作用通路的6个基因中,有2个氧增强蛋白基因(OEE2、OEE3)、2个光系统Ⅱ蛋白基因(PSBR、PSB27)、2个光系统Ⅰ蛋白基因(PSAF、PSAK);氮代谢通路的3个基因包括2个硝酸还原酶基因(NR)和1个亚硝酸还原酶基因(NiR)(表5)。利用荧光定量PCR对光合作用通路和氮代谢通路的9个基因分别在根和叶片中进行表达分析,结果如表5所示,9个基因在根和叶片中的荧光定量PCR与RNA-seq的表达分析结果具有相同的变化趋势,说明RNA-seq的表达分析结果具有较高的准确性;在氮素响应过程中,9个基因在根和叶片中具有相似的表达模式,基因表达量随着氮素浓度的升高而升高,缺氮处理中表达量最低,高氮处理中表达量最高;在每一种氮素浓度处理中,光合作用通路的6个基因在叶片中的表达量均高于在根中的表达量。
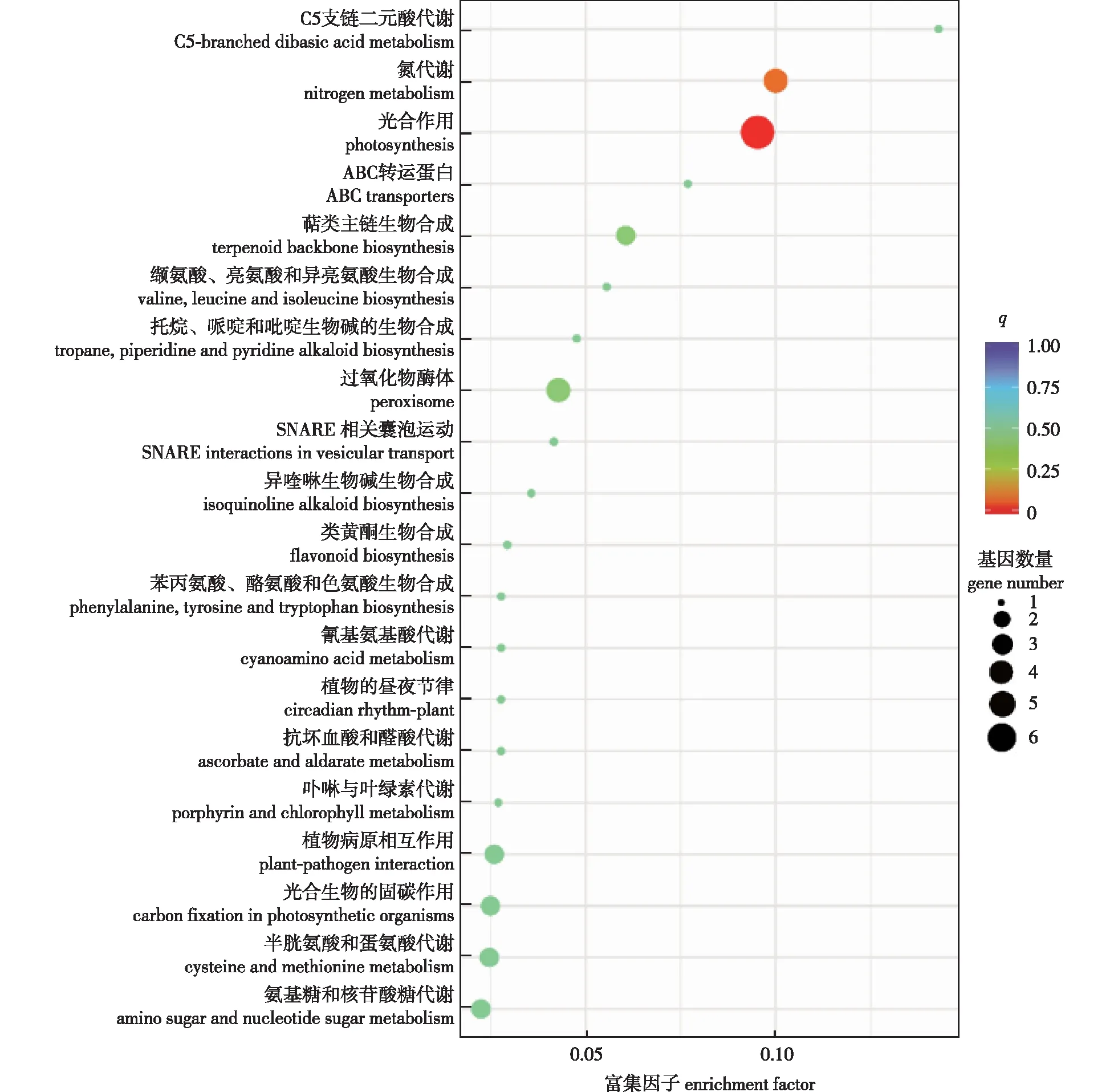
图3 差异表达基因的KEGG分析Fig. 3 KEGG analysis results for differentially expressed genes

表5 9个差异表达基因在根和叶片中的表达量
3 讨 论
笔者在前期的研究中发现青檀在施加不含有NH4NO3的Hocking’s营养液的缺氮条件下与正常有氮条件下相比,地上部分生长量显著降低;在分别施加2、5、10 mmol/L NH4NO3的Hocking’s营养液的中氮条件下,地上部分生长量变化不显著;在分别施加20、30、40、50 mmol/L NH4NO3的Hocking’s营养液的条件下,地上部分生长量逐渐增加;在施加浓度超过50 mmol/L NH4NO3的Hocking’s营养液时,青檀地上部分生长量开始降低。本研究选取0、2、50 mmol/L NH4NO3的Hocking’s营养液分别作为缺氮(N0)、中氮(N2)、高氮(N50)处理条件,发现缺氮处理促进了青檀根系生长,抑制了地上部生长,抑制了光合作用,而高氮处理抑制了根系生长,促进了地上部生长,促进了光合作用,此结果与他人在杨树中的研究结果相似[5]。植物在形态发育和生理方面对环境条件变化做出的响应是受到基因调控的,青檀作为没有全基因组测序数据的非模式植物树种,研究其基因表达模式最高效的手段是进行转录组测序。
本研究中,笔者对青檀进行3代全长转录组测序,通过数据分析得到11 801 条基因,其中11 232(95.18%)条基因得到相应的注释。根系是获取土壤中营养物质的主要器官,并且通过改变细根的长度和质量来响应土壤养分的变化,于是,本研究对不同氮素水平N0、N2、N50的3个生物学重复的根系样本取材,总共构建了9个文库,进行2代转录组测序,数据与3代测序结果进行比对,表达谱分析的结果显示氮素水平差异越大,产生的差异表达基因数量就越多,N50比N2与N2比N0上调差异表达基因集合交集的76个基因和下调差异表达基因集合交集的32个基因全部属于N50vs N0上调和下调差异表达基因集合,说明这76个基因随着氮素水平提高稳定上调表达,32个基因随着氮素水平提高稳定下调表达;也说明这些基因在各自的代谢通路中被强烈激活或抑制表达,暗示与氮素响应存在密切关系。同时,在本研究中,青檀的地上部分生长量随着氮素浓度提高而提高,根系生长量随着氮素浓度提高而降低。所以,笔者重点对这108个在3种氮素水平稳定表达上调或下调的差异表达基因进行研究。通过KEGG通路富集分析发现,排在前两位的分别是光合作用和氮代谢,富集的基因数量分别是6和3。6个光合作用通路基因中包括2个氧增强蛋白基因、2个光系统Ⅰ蛋白基因、2个光系统Ⅱ蛋白基因。3个氮代谢通路基因包括2个硝酸还原酶基因和1个亚硝酸还原酶基因。
氧增强蛋白2和3是叶绿体类囊体膜的放氧复合体的2个外周蛋白,表观分子质量分别是23和17 ku,在光系统Ⅱ中起到维持放氧活力的作用,水稻的氧增强蛋白2基因在盐胁迫中表达上调[20],拟南芥的氧增强蛋白2在病原体胁迫过程中行使功能[21]。光系统Ⅱ和光系统Ⅰ蛋白基因通常认为主要在叶片中行使功能。拟南芥和陆地棉的研究发现光合作用通路基因在非光合作用器官根中发生表达,但表达量低于在叶片中的表达量[22-23]。玉米(Zeamays)叶片的光合作用通路基因在低氮水平表达下调,在高氮水平表达上调[24]。低氮处理时,西瓜(Citulluslanatus)叶片中的多个光系统Ⅰ蛋白基因和光系统Ⅱ蛋白基因大幅下调,而在根中没有检测到任何光合作用通路基因的表达[25]。杨树(Populusspp.)叶片的2个光系统Ⅰ蛋白基因、3个光系统Ⅱ蛋白基因在中氮比缺氮和高氮比中氮中都是差异表达基因,且这5个基因表达模式相似,在缺氮处理中表达量最低,在中氮处理中表达量最高[5]。青檀与作为木本模式植物的杨树在此方面出现较大差异。通常情况下,光合作用通路基因在叶片中具有高于其他营养器官的表达水平,本研究中,受到氮素浓度变化的影响,光合作用通路的6个基因在光合作用的主要器官叶片中发生了与根中相似的明显的表达趋势变化,并且在叶片中的表达量高于根中的表达量,推测氮素浓度变化导致光合作用通路的6个基因在根和叶片中均发生了表达变化,与光合作用基因表达量随着氮素浓度的升高而升高相一致的是高氮素水平明显促进了光合作用,导致了叶绿素a、叶绿素b、类胡萝卜素含量的提高,增强了净光合速率和气孔导度,而光合作用的增强促进了青檀地上部生长。
硝酸根需要还原为铵根才能被植物体利用,硝酸还原酶是硝酸根同化过程中的限速酶,亚硝酸还原酶是一个关键控制酶,两种酶偶联调节完成硝酸根的同化。西瓜在用低氮和高氮处理时,叶片中4个硝酸还原酶基因和1个亚硝酸还原酶基因是高氮比低氮的表达差异基因,并且在高氮处理时表达量比低氮大幅提高,并且西瓜的地上部生长随着氮素浓度升高而提高[25]。杨树根的1个硝酸还原酶基因在中氮比缺氮和高氮比中氮中都是差异表达基因,且在缺氮处理中表达量最低,在高氮处理中表达量最高[5]。杨树叶片的1个亚硝酸还原酶基因在中氮比低氮中是差异表达基因,且在中氮处理的表达量比低氮大幅下降[5]。一般情况下,硝酸盐能够诱导硝酸还原酶基因和亚硝酸还原酶基因的表达,从而导致硝酸还原酶和亚硝酸还原酶活性的提高,使硝酸根的同化效率得到提高。本研究结果显示,青檀的硝酸还原酶基因和亚硝酸还原酶基因在氮响应过程中具有与木本模式植物杨树基因不同的作用方式,在不同氮素水平,在青檀的根中,2个硝酸还原酶基因和1个亚硝酸还原酶基因都在缺氮处理中表达量最低,在高氮处理中表达量最高,并且在根和叶片中具有一致的表达趋势,暗示随着氮素浓度的升高,硝酸还原酶和亚硝酸还原酶在根和叶片中的同化能力得到了提高。推测随着氮素浓度的升高,硝酸还原酶基因和亚硝酸还原酶基因表达上调,同时,硝酸还原酶和亚硝酸还原酶活性得到相应提高,提高了硝酸根的同化效率,同化产物大多运输到地上部被同化利用,而根系生长得到抑制。
本研究用0、2、50 mmol/L NH4NO3的Hocking’s营养液对青檀进行了氮素处理试验,研究发现缺氮处理促进了青檀根系生长,抑制了地上部生长,抑制了光合作用,而高氮处理抑制了根系生长,促进了地上部生长,促进了光合作用。研究发现76个基因随着氮素水平提高稳定上调表达,32个基因随着氮素水平提高稳定下调表达。通过KEGG对这108个差异表达基因进行功能富集分析,结果表明光合作用通路和氮代谢通路显著富集,其中光合作用通路包含6个基因(OEE2、OEE3、PSBR、PSB27、PSAF、PSAK),氮代谢通路包含3个基因(2个NR和1个NiR)。表达分析结果说明这9个基因在根和叶片中具有相似的表达模式,基因表达量随着氮素浓度的升高而升高,缺氮处理中表达量最低,高氮处理中表达量最高,暗示这9个基因在青檀氮响应过程中具有重要功能。笔者推测随着氮素浓度的提高,光合作用通路基因表达上调,促进了光合作用,硝酸还原酶基因和亚硝酸还原酶基因表达上调,提高了硝酸根的同化效率,最终造成青檀地上部生长得到加强,根系生长受到抑制。在目前没有参考基因组的情况下,本研究为探讨青檀氮素响应分子机理、发掘关键候选基因提供了有力依据。
猜你喜欢
中国农业大学学报(2022年3期)2022-05-19
土壤学报(2022年1期)2022-03-08
昆明医科大学学报(2021年5期)2021-07-22
文萃报·周二版(2019年23期)2019-09-10
天津诗人(2018年4期)2018-11-14
商情(2018年5期)2018-03-28
文苑·经典美文(2016年8期)2016-05-14
湖北农业科学(2014年8期)2014-08-08
中国酿造(2014年9期)2014-03-11
生物加工过程(2013年1期)2013-03-11
