
TRPV4在青光眼性视神经病变中的研究进展
2023-12-02 06:20:06周亚莎彭清华
国际眼科杂志 2023年12期
黄 雨,夏 鑫,周亚莎,彭清华,2
0 引言
青光眼(glaucoma)是全球发病率排名首位的不可逆性致盲眼病,其致盲性与治疗难点均在于该病所引起的视神经病变。近年来,针对于青光眼性视神经病变(glaucomatous optic neuropathy,GON)的发病机制进行了大量研究,并形成了多种机制学说,包括机械压力学说、局部血管缺血学说、自身免疫异常学说、氧化应激学说、谷氨酸的神经兴奋性毒性作用、一氧化氮(nitric oxide,NO)的视神经毒性反应、基因突变与遗传等[1]。但其中任何一种机制学说均无法解释GON所有类型的发病机制,如机械压力学说不能对正常眼压性青光眼(normal-tension glaucoma,NTG)所导致的视神经病变作出解释,故在临床上,单纯的采取降眼压措施无法治疗NTG。鉴于此现状,寻找一种针对于GON所有类型的共同发病机制或共同靶点,将有助于该病的诊疗。
近几年,瞬时受体电位通道香草酸亚家族4(transient receptor potential channel vanillic acid subfamily 4,TRPV4)受到了密切关注,且有研究发现TRPV4在视网膜中广泛分布,其持续激活可导致视网膜神经节细胞(retinal ganglion cells,RGCs)死亡[2],而GON最核心的病理改变即为RGCs死亡[3],故TRPV4是否可将GON的多种发病机制联系起来,值得关注。本文将对TRPV4及其在GON多种发病机制中所发挥的作用作一综述,以期为该病的基础研究和临床诊疗提供参考依据。
1 TRPV4概述
1.1TRPV4的结构TRPV4属于瞬时受体电位通道(transient receptor potential,TRP)超家族的成员之一,是一种典型的非选择性阳离子通道。其在人类各组织细胞中的表达,是以TRPV4基因编码,且由871个氨基酸组成。TRPV4蛋白的构造是一种四聚体结构,有功能的TRPV4具有6个跨膜片段(S1~S6)和4个亚基。对于跨膜片段,其主要作用区域集中在S5和S6之间的离子通道孔,该孔可保障阳离子的顺利通过,如钙离子(Ca2+)等[4]。TRPV4所包含的氨基端(N)和羧基端(C)均朝向细胞质侧,具有多种不同的功能区域。如N末端拥有6个锚蛋白重复序列(ankyrinrepeat,ANK),这些ANK构成的双螺旋结构可作为TRPV4磷酸肌醇结合位点(phosphoinositide-binding site,PIBS),该位点可感知各种物理或化学刺激,从而调节TRPV4发挥作用;C末端具有的功能域主要包括钙调蛋白结合位点、细胞骨架蛋白结合位点等,这些位点将有助于细胞外物质通过TRPV4调节具有该通道的细胞内物质改变,从而发挥不同效应[5]。TRPV4结构见图1[6]。
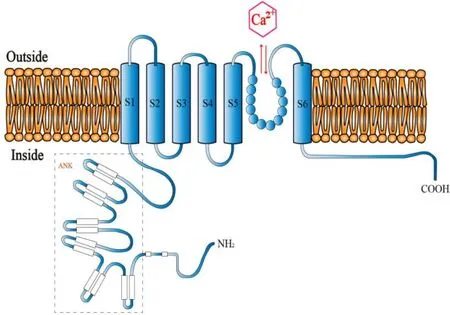
图1 TRPV4结构图。
1.2TRPV4的功能TRPV4广泛分布于各组织脏器中,在脑、心脏、肺脏、脉管组织中均有表达。迄今为止,研究已证实TRPV4可被各种物理及化学刺激激活,包括温度、渗透压、机械压力、血流切应力、炎症因子、氧化应激产物等,从而参与各系统器官疾病的发生发展[7]。在高血压、肺动脉高压等心血管疾病研究中发现,经TRPV4蛋白通道调节Ca2+内流量可引起血管收缩和舒张[8]。在克罗恩病、溃疡性结肠炎等炎症性肠病研究中发现,TRPV4介导的Ca2+信号传导通路可被炎症因子激活,参与进一步释放促炎细胞因子、激活免疫细胞和增加血管通透性的过程[9]。在心肌缺血再灌注损伤、充血性心力衰竭等心肌损伤性疾病的研究中发现,该类损伤模型中TRPV4蛋白表达明显上调,细胞内Ca2+浓度增加,引发抗氧化酶活力下降,氧化产物增多,且细胞内线粒体功能发生紊乱,从而造成心肌细胞损伤[10]。另外,在对顽固性癫痫、创伤性脑损伤等神经系统疾病的研究中发现,出血、缺氧、炎症及先天发育不良等一系列诱因,均可上调脑组织中TRPV4蛋白的表达,进而改变神经元活性,破坏血脑屏障,诱导相应脑组织细胞损伤[11]。归纳TRPV4所引发的病理改变,其主要来源于对Ca2+细胞内外的调节。根据其具体的分布位置,感受相应刺激后,可作出不同的机制应答。正是因为TRPV4可介导不同机制的发生,从而使TRPV4在具有多种致病机制的疾病研究领域深受关注。
2 TRPV4在GON中的作用
2.1机械压力传统意义上的机械压力学说,并不能解释所有类型的GON。其强调病理性高眼压(pathological high intraocular pressure,ph-IOP)对视盘乃至整个视网膜的直接压迫,阻断RGCs细胞体与大脑之间的信号传递,对视神经摄取神经营养因子(neurotrophic factor,NF)起到抑制作用,同时还可减少甚至中断视乳头(optic nerve head,ONH)及其周围组织的血流供应,从而引起视神经损伤及RGCs死亡[12]。但上述原理并不能对NTG这一类型的发病机制作出解释。NTG发病过程中,虽然具有GON的表现,但与ph-IOP并无直接关系。近年来,关于NTG的研究发现,眼内压(intraocular pressure,IOP)与颅内压(intracranial pressure,ICP)之间的压力差对该类型的视神经损伤发挥着重要作用,即当IOP处于正常数值范围内时,由于过低的ICP会导致跨筛板压力差升高,而此时尽管IOP处于正常数值范围内,同样也会对视神经造成损伤[13]。另外,视神经压力敏感性也可对视神经产生影响。视神经压力敏感性是指视神经对机械压力的耐受性,敏感性越低,视神经对机械压力的耐受性则越高,反之亦然。当压力敏感性降低时,即使IOP超出正常范围,对视神经也不会构成威胁,即为高眼压症。反之,当压力敏感性升高时,即使IOP处于正常范围,也会对视神经造成损伤,即为NTG[14]。故最新观点认为,机械压力导致GON的原理,不单纯只是IOP升高,可能还与ICP改变、视神经压力敏感性升高或三者之间的相互作用有关。
TRPV4作为一种非选择性力敏感离子通道,可感受细胞膜表面应力变化,与机械压力介导的相关疾病机制密切相关。分布于视网膜中RGCs的TRPV4,可被ph-IOP机械刺激激活,介导Ca2+、Na+等阳离子流入细胞内,增加RGCs的放电率,并在TRPV4持续激活后,促使线粒体过度代偿性耗竭,导致RGCs死亡,参与由于ph-IOP所致视神经损伤的发病过程[15]。分布于小梁网、睫状体等部位的TRPV4,则可对房水的生成产生调控作用。研究发现,小梁网细胞中TRPV4激活可引起应力纤维重塑和纤维连接蛋白上调,使房水流通途径阻力增加,进而形成ph-IOP[16]。而睫状体非色素睫状上皮细胞(non-pigment epithelium,NPE)中TRPV4高表达,不仅可介导NPE外Ca2+内流,引起房水的大量产生,升高IOP[17],也可使NPE外褪黑素水平升高,而褪黑素可减少睫状上皮氯化物的产生,以降低IOP[18]。此外,TRPV4的激活还可提高视神经压力敏感性,降低RGCs对机械压力的耐受性,引起视神经缺血、缺氧,进而引起GON的发生[19]。从上述研究来看,存在于视网膜中RGCs及眼部其他部位的TRPV4,其介导的机械压力机制可以解释ph-IOP及NTG中提出的视神经压力敏感性升高所引起的GON发病机制。因此,TRPV4可被视为GON发病机制中机械压力作用的重要靶点。
2.2血管缺血随着血管成像技术在眼科中的广泛应用,大量临床工作者发现视网膜及ONH的局部血液供应障碍是GON患者的重要体征之一。在临床上,激光血流散斑成像(laser speckle flowgraphy,LSFG)和光学相干断层扫描血管成像(optical coherence tomography angiography,OCTA)提供了大量局部血管低血流量是造成GON重要因素的证据。有研究者运用LSFG对视野前期青光眼(preperimetric glaucoma,PPG)患者的ONH局部血流进行监测,发现PPG患者的ONH局部血流量明显下降,且ONH结构损伤与血流量的改变密切相关[20]。而在OCTA的运用上,则发现不同类型的青光眼患者局部视网膜深、浅部血管丛密度均明显降低[21]。基础研究发现,眼压慢性轻中度升高的动物模型视神经初期血流量会代偿性增加,视网膜神经纤维层(retinal nerve fiber layer,RNFL)厚度无明显减少,而后随着时间的推移,其血流量会呈现出大幅度下降,并伴有大量RNFL及RGCs丢失[22]。另外,鉴于血管遍布全身的特性,与血管供血不足相关的全身其他系统疾病也可影响眼部血液供应,进而导致视神经损伤,如在NTG的研究中发现,偏头痛、Flammer综合征(原发性血管调节障碍)等其他系统疾病均与NTG的发病有关[23]。总之,眼局部血液循环障碍与ph-IOP一样,属于GON发生发展的重要危险因素之一。
TRPV4在血液循环过程中所发挥的作用主要是对血管的收缩与舒张进行调节。关于冠状动脉微血管功能障碍的研究发现,TRPV4可介导Ca2+进入内皮细胞,增强冠状动脉微血管的舒张[24]。而在人视网膜微血管内皮细胞(human retinal capillary endothelial cells,HRCECs)中存在的TRPV4也可调控细胞外Ca2+内流,发挥HRCECs迁移和新生血管形成的作用,进而参与多种视网膜血管疾病的发病进程[25]。同时,激活的TRPV4还可改变HRCECs之间的紧密连接程度,调节血-视网膜屏障的通透性及视网膜深、浅层血管血流量的大小[26]。关于视网膜缺血再灌注损伤的研究发现,有效抑制TRPV4表达可减少损伤后RGCs凋亡及新生血管形成[27]。另外,运用具有活血化瘀功效的中药干预青光眼模型小鼠发现,该类药物可有效降低视网膜中TRPV4蛋白表达,进而发挥保护视神经及RGCs的作用[28],而现代研究认为中医中活血化瘀药主要是发挥改善局部血液循环的作用[29],故可间接证明TRPV4可通过调节视网膜及ONH局部血液循环,对GON进行改善。结合临床上对GON患者采取改善局部血液微循环措施的有效性及TRPV4对微血管收缩与舒张的调节作用,推测TRPV4可作为GON血管缺血机制中的重要蛋白调控位点。
2.3自身免疫异常随着对GON研究的深入,自身免疫异常被认为是GON发病的又一重要机制。有研究者发现,在青光眼实验模型中,损伤的视神经周围均存在巨噬细胞、T细胞浸润,且RGCs及其轴突逐渐丢失与该类炎症细胞的浸润程度具有直接关系[30]。此外,发生GON时,各种致病因素可造成视网膜中胶质细胞过度活化,继而产生大量细胞因子,加重局部炎症反应。如当发生IOP增高、局部轴突信号运输障碍等异常情况时,可使A1型星形胶质细胞、M1型小胶质细胞过度活化,引起视网膜及ONH微环境物质改变,包括促炎因子的生成和释放等,最终导致RGCs存活率降低[31];A2型星形胶质细胞、M2型小胶质细胞则可上调抗炎介质和生成NF,参与局部组织修复[32]。而Müller细胞在青光眼发病早期,可通过增加谷氨酸摄取、分泌抗氧化物质和NF,对RGCs起到保护作用;随后由于对该细胞的过度激活,其分泌的一系列促炎因子可对视神经起到损伤作用[33]。除眼局部存在炎症反应的相关证据外,全身免疫异常也与GON的发病具有相关性,如在GON患者中,其外周血巨噬细胞所分泌的核苷酸结合寡聚化结构域样受体蛋白3(nucleotide-binding oligomerization domain-like receptor protein 3,NLRP3)炎症复合体,包括NLRP3、凋亡相关微粒蛋白(ASC)、半胱氨酸天冬氨酸蛋白酶1(caspase-1),与视神经损伤程度存在显著的正相关性[34]。
TRPV4在多种免疫细胞中均存在表达,可被机械压力、温度依赖性刺激及炎症反应中所产生的细胞因子等激活,其活化可参与多种炎症性疾病发生发展的过程。Sun等[35]关于骨性关节炎的研究发现,抑制TRPV4的表达可通过活性氧(reactive oxygen species,ROS)/NLRP3信号通路有效降低滑膜中M1巨噬细胞的浸润,减轻局部炎症所引起的病理改变。而有研究发现青光眼模型大鼠视网膜中的TRPV4持续激活,可通过JAK2/STAT3/NF-κB信号通路诱导Müller细胞胶质化,释放大量细胞因子,同时引起肿瘤坏死因子-α(tumor necrosis factor-α,TNF-α)升高,加剧视网膜中RGCs凋亡[2]。在成年猪视网膜体外培养过程中,研究者特异性抑制该部位的TRPV4表达,发现可有效增强RGCs存活率,其原理主要与减弱胶质细胞反应,维持免疫稳态有关[36]。此外,在其他眼部疾病的研究中,同样也可观察到视网膜TRPV4与炎症反应密切相关。如针对视网膜脱离(retinal detachment,RD)的实验发现,RGCs的死亡与Müller细胞表面存在的TRPV4激活有关,其具体机制为TRPV4表达上调,刺激Müller细胞生成单核细胞趋化蛋白1(monocyte chemoattractant protein-1,MCP-1)等炎性细胞因子,从而对视网膜产生不利影响[37]。从以上研究来看,视网膜中TRPV4的表达可通过调控炎症反应参与GON的发病过程。
2.4氧化应激关于青光眼的一系列临床及基础实验发现,诸多眼局部组织存在氧化应激(oxidative stress,OS)标记物,其中包括视神经及其周围血管等组织,且运用抗氧化药物干预后,可对视网膜中RGCs起到保护效应,表明OS可能在GON的病变过程中发挥作用[38-39]。Edwards等[40]运用泛醇(一种以辅酶Q10为主要活性成分的抗氧化剂)对青光眼DBA/2J小鼠模型进行干预,发现该抗氧化剂可通过增加转录因子A和氧化磷酸化复合物蛋白减少凋亡蛋白的表达,以起到提高RGCs存活率的作用。与此同时,有研究证实ROS可使小梁网细胞功能受损,房水循环动力学异常,进而引起ph-IOP的发生,机械压迫视神经,导致RGCs凋亡[41]。另外,线粒体功能障碍已被明确是GON发生的重要一环。Hass等[42]研究发现,通过敲除线粒体解偶联蛋白,减少氧化蛋白修饰,增加视网膜组织中的有丝分裂水平,促进线粒体功能增强,可达到减少青光眼小鼠模型中RGCs死亡的目的。而线粒体作为产生ROS的主要场所,其功能障碍可导致GON的发生,故推测GON的发生理论上可能与ROS的改变有关。
TRPV4的表达可引发机体组织中OS反应,这一事实已在大量疾病研究中得以验证。其途径主要有三种:(1)TRPV4被激活后,细胞外Ca2+大量内流,细胞内钙蛋白酶随后被其激活,引发黄嘌呤脱氢酶转变成黄嘌呤氧化酶,进而导致ROS等自由基数量增加;(2)细胞内Ca2+浓度增加可激活Ca2+依赖性磷脂酶A2,从而使花生四烯酸增多,在环氧合酶和脂加氧酶的作用下产生大量氧自由基;(3)细胞内Ca2+增加可引起过氧化氢酶(catalase,CAT)、谷胱甘肽过氧化物酶(glutathione peroxidase,GSH-Px)等抗氧化酶活性下降,进而使机体抵御氧化损伤的能力下降[43]。研究发现,在病理状态下可通过激活TRPV4抑制CAT、GSH-Px及增加一氧化氮合酶(nitric oxide synthase,NOS)的活性,以达到OS总效应增强的作用,从而对神经细胞造成破坏[44]。Özimek等[45]运用褪黑素对缺氧诱导的损伤细胞进行干预,发现褪黑素可通过抑制TRPV4激活继而降低线粒体自由活性氧数量和脂质过氧化状态,提高GSH-Px活性和细胞总抗氧化能力,从而发挥细胞保护作用。那么,存在于眼局部的TRPV4是否同样也可通过OS反应对视神经及RGCs造成损伤呢?针对于TRPV4在视网膜及视神经局部病变所发挥的OS作用,并没有相关文献研究进行支撑。现阶段只能通过TRPV4与OS之间的关系间接证明在青光眼发病过程中TRPV4可通过OS反应对视神经等眼局部组织造成损伤。故有关通过调控视网膜等眼局部组织的TRPV4表达降低OS反应,以此发挥保护RGCs作用的直接证据还需要进行深入研究。
2.5其他除上述机制外,谷氨酸与NO对视神经的毒性反应,在GON发病过程中也起着重要作用。正常生理状态下,谷氨酸可对人眼视觉的形成与发育发挥积极效应。但当发生GON时,涉及该病发生的多条途径,包括ph-IOP、局部组织缺血缺氧、炎症反应的激活等,均可造成谷氨酸大量堆积,激活N-甲基-D-天冬氨酸受体(谷氨酸受体),使细胞外Ca2+大量内流,从而对视神经产生兴奋性毒性反应[45]。而NO在眼局部组织中主要由NOS氧化后生成,对青光眼所致眼组织损伤具有保护作用,其内源性释放可促进房水引流、调节眼血流量、维持IOP稳态。但当NOS/NO系统失调,大量NO生成,可同时产生多种自由基,如过氧亚硝酸盐,引发局部组织缺血、神经元变性等效应,从而对GON的发生发展产生恶性循环[46]。由于谷氨酸、NO对视神经及RGCs产生损害效应是基于上述4种机制过程,故理论上TRPV4激活可通过影响谷氨酸及NO的数量进而造成GON的发生。当然,该推论仍需相关研究进行验证。
3 小结
由于现阶段对GON发病机制认识的多样化,使得在临床治疗该病的过程中不具备针对性,且现代化医学技术还无法对青光眼所造成的视神经损伤进行恢复,故寻找在GON发病过程中多种机制的“连接点”,将有助于该病的早期诊断和治疗。本文对GON的几个重要发病机制进行了阐述,并结合近年来关于TRPV4在视网膜中表达的相关研究,发现TRPV4的激活的确可通过GON的多条发病途径对视神经及RGCs造成损伤。那么,通过本文综述,有关作用于TRPV4的药物是否能够针对性治疗GON的所有类型,值得进一步探索,这也将为该病的新药研发提供新思路。
猜你喜欢
中老年保健(2022年3期)2022-08-24 02:57:52
现代仪器与医疗(2022年2期)2022-08-11 09:53:56
基层中医药(2021年8期)2021-11-02 06:24:54
基层中医药(2021年6期)2021-11-02 05:46:04
中医眼耳鼻喉杂志(2021年1期)2021-07-22 07:38:28
中医眼耳鼻喉杂志(2021年2期)2021-07-21 08:53:34
眼科学报(2021年6期)2021-07-18 02:06:02
科学(2020年3期)2020-11-26 08:18:30
中医眼耳鼻喉杂志(2019年3期)2019-04-13 05:26:50
湖南中医药大学学报(2016年1期)2016-12-01 04:08:18
