
Effect of chaperone–client interaction strength on Hsp70-mediated protein folding
2023-12-02 09:22LujunZou邹禄军JiajunLu陆伽俊andXiulianXu徐秀莲
Chinese Physics B 2023年11期
Lujun Zou(邹禄军), Jiajun Lu(陆伽俊), and Xiulian Xu(徐秀莲)
1Department of Physics,National Laboratory of Solid State Microstructure,Nanjing University,Nanjing 210093,China
2School of Physics Science and Technology,Yangzhou University,Yangzhou 225002,China
Keywords: protein folding,molecular chaperone,molecular dynamics,Hsp70
1.Introduction
Correct folding is a prerequisite for most proteins in order to perform their biological functions.[1]Although natural foldable proteins have well evolved to shape up a funneled energy landscape,[2–5]functional requirements,[6–10]and crowding environment[11]often impose additional frustration to the landscape.Consequently, protein folding in cellular environment may encounter the risk of misfolding and irreversible aggregation, which can be closely relevant to a number of severe diseases, such as Alzheimer’s disease, Parkinson disease,type-II diabetes,etc.[12]Cells utilize various chaperones to protect proteins from misfolding.[13]
Hsp70 is one of the most widely investigated molecular chaperones and it is ubiquitous in both prokaryote and eukaryote.[14,15]It consists of an N-terminal ATP binding domain (NBD) and a C-terminal substrate binding domain(SBD).The ATP binding and hydrolysis drive the conformational cycle of the Hsp70 SBD between a closed conformation (Fig.1(b)) and an open conformation (Fig.1(c)).[16]The closed conformation has a higher affinity with substrate(client) protein, whereas the open conformation has a lower affinity to substrate.[16,17]The substrate protein can bind to and release from the open conformation of the Hsp70 SBD.Through such Hsp70–client interactions during the conformational cycle of the Hsp70, the Hsp70 chaperone can either protect unfolded conformations from aggregation by acting as a holdase, or actively convert the misfolded substrate protein to unfolded conformations by acting as a unfoldase.[18–20]Recent computational studies by Lu and coworkers revealed that the interactions between substrate and Hsp70 at open conformation are essential for protein folding, and the Hsp70 tends to accelerate protein folding by remodeling its folding energy landscape,which therefore suggests a foldase mechanism.[21]However,how the interaction strength between Hsp70 and the substrate protein affects protein folding was not well studied,which is crucial for understanding the sequence-function relation of proteins and therefore for protein design.

Fig.1.Structure of chaperone and substrate.(a)Cartoon representation of the native structure of the substrate hTRF1.The native contact are formed mainly between helices H1 and H2 and between helices H1 and H3.The hydrophobic patch for binding the SBD of the Hsp70 is colored by bright orange.Panels(b)and(c)are representative structures of the Hsp70(SBD)-substrate complex at the closed(b)and open states(c).
In this work, we investigate the effect of chaperone–client interaction strength on the Hsp70-mediated folding of a model protein hTRF1 by using a structure-based coarsegrained (CG) protein model within the framework of energy landscape theory.[3,8,21]The chaperone binding site of this protein was identified in previous work.[22,23]In addition,hTRF1 is marginally stable, and the chaperone binding site with hydrophobic residues can be exposed in the transiently unfolded conformation, which makes it have the potential of misfolding under crowding environment.This protein has also been used as the model system to investigate the chaperonemediated protein folding mechanism in previous experimental work.[22,24]We showed that the folding rate of hTRF1 has a non-monotonic dependence on the interaction strength.There is an optimal chaperone–client interactions strength at which the chaperone has the highest efficiency of assisting the substrate protein folding.The current results demonstrate the importance of the chaperone–client interaction strength in understanding the chaperoning function of Hsp70.
2.Methods
2.1.Coarse-grained protein model
Molecular dynamics simulations have been widely used in the studies of protein folding,protein aggregation,and other complex processes.[25–27]For large systems involving the time scale longer then microsecond,coarse-grained models are often useful due to its high sampling efficiency.[28–30]In this work, we used a coarse-grained model to describe the folding of substrate protein and the conformational changes of the molecular chaperone.In the coarse-grained model, each spherical bead represents a residue and it locates at thecαposition.The energy function of CG model is given by
whereRcollectively represents the coordinates of coarsegrained beads in a given structure andR0represents the coordinates of the reference native structure.For the substrate protein hTRF1, the reference native structure coordinatesR0were obtained from Protein Data Bank with the entry number 1BA5.[31,32]Vb(R|R0)represents the potential energy function for a virtual bond.(R)is a generic flexible local potential extracted based on statistical survey of coiled segments of the structures in Protein Data Bank.[33,34]VSB(R|R0)corresponds to the structure-based potential shaping up a funnelled energy landscape.[3,4,35,36]We used the AICG2+ model, which is a structure-based energy function optimized by using a multiscale strategy to describe the funnelled energy landscape of protein folding.[7,8,34,37,38]VKH(R) is introduced to describe the interactions of the residue pairs that do not form direct contacts in the native structure and is a knowledge-based nonspecific statistical potential.[39]Vex(R)describes the excluded volume effect of the non-native residue pairs.[33]The last term describes the electrostatic interactions between the charged residue pairs,which is given by the Debye–H¨uckel theory.[40]
On the top of the AICG2+ energy function with minimal frustration,we also introduced an additional term to consider the ruggedness of the energy landscape following previous work,[21]which is essential for the reasonable description of the misfolding of the substrate protein.As demonstrated by the NMR structure of hTRF1(shown in Fig.1(a)), the native contacts of the hTRF1 are formed mainly betweenαhelix 1(H1) andαhelix 2 (H2), as well as betweenαhelix 1 (H1)andαhelix 3 (H3).However, there are rare native contacts between H2 and H3.Consequently,we introduced non-native contacts between H2 and H3.The interactions of the nonnative contacts are described by the following Lennard–Jones potential:
whererijrepresents the distance between residuesiandj,σ=5 ˚A is the equilibrium distance,and non-native contacting strengthε=3.4 kcal/mol characterizes the frustration extent of the energy function.
Hsp70 conducts its chaperone function by switching the conformation of SBD between its open and closed states,which is governed by an allosteric mechanism coupled with ATP binding and hydrolysis.[16,17]Following previous work,we constructed a double-basin energy function to describe the intramolecular motions of Hsp70 SBD,[21,41]which is given by the following equation:
In the formula mentioned above, ‘s’ represents the ATP and ADP states of Hsp70.The open and closed structures of the Hsp70 are utilized to represent the reference coordinates, which were obtained from the Protein Data Bank with entry numbers 4B9Q and 4EZW,respectively.[32,42]The parameters ∆Vsand∆sregulate the relative stability and barrier height between the two basins.By tuning these parameters, one can roughly reproduce the experimental observation that Hsp70 predominantly adopts a closed conformation when binding with ADP and an open conformation when binding with ATP.[21,43]By switching the double-basin energy function from ADP state to ATP state, it is possible to reasonably simulate the conformational transition of Hsp70 SBD from closed state to open state.[16–18,21]Similar energy function with double-basin topography has been successfully used in describing the ligand binding coupled folding and conformational motions of allosteric proteins,[28,41,44]the molecular mechanism of enzyme catalysis,[9,45,46]and filament assembly[47]in previous works.The energy function describing the hydrophobic interaction between the chaperone and substrate is given by[38,48]
The first term accounts for the hydrophobic interactions between the chaperone and substrate.It involves all residues in the chaperone and in the binding site of the substrate protein.The substrate residues contributing to the chaperone binding are K28 to H32 in H2 of hTRF1.[22]The parameterCHPregulates the strength of hydrophobic interactions, enabling us to explore the role of intermolecular interactions between the chaperone and the substrate protein on chaperone-mediated protein folding.The second term represents the excluded volume interactions.More details of energy function and parameters can be found in previous works.[21,48]
2.2.Molecular simulation and data analysis
The molecular simulations were performed using the software CafeMol 3.0 with langevin thermostat at temperature of 300 K.[48]Prior to the folding simulations,we conducted equilibrium simulations with the substrate prebound to the closed state of Hsp70 for 2×108MD steps to prepare the unfolded ensemble of the substrate.Then we switched the double-basin energy function of chaperone from ADP state to ATP state,leading to the opening of the Hsp70 SBD.Subsequently, we simulated the folding and release of substrate at the open conformation of the Hsp70 SBD.
Following previous work,we constructed the reaction coordinatesQ,Q12,Q13,N23, andNmisto characterize the substrate folding.[21]The reaction coordinateQdescribes the fraction of formed native contacts.Similarly,Q12andQ13represent the fractions of the formed native contacts between H1 and H2, and between H1 and H3, respectively.N23is calculated as the total number of possible contacts between H2 and H3, andNmisis the number of non-native contacts formed between H2 and H3.To describe the dissociation process between hTRF1 and Hsp70, we have defined reaction coordinatesNHPandRSP.NHPrepresents the number of formed contacts between Hsp70 SBD and the H2 of the substrate protein hTRF1.RSPrepresents the distance between the substrate binding pocket of Hsp70 SBD and the hydrophobic binding site on H2 of hTRF1.The substrate is considered to be released from the molecular chaperone if the inter-molecule interaction energy is equal to or larger than 0.The two-dimensional free energy landscape is calculated byF(x1,x2) =-lnP(x1,x2), wherex1andx2are the reaction coordinates and theP(x1,x2) is the probability distribution alongx1andx2.Following previous work, we defined five distinct conformational states, i.e., the native state N (Q ≥0.8)), the unfolded state U (Q12< 0.3,Q13< 0.3,Nmis=0), the misfolded stateI23(Nmis≥1), the on-pathway intermediate stateI13(I13:Q13>0.8,Q12<0.3,Nmis=0),and the on-pathway intermediate stateI12(Q13<0.3,Q12>0.8,Nmis=0), respectively.[21]The folding pathway is composed of a series of the above defined states in the trajectory.The structures of Hsp70 and hTRF1 are visualized by PyMOL and VMD.[49,50]
3.Results and discussion
Simulating the whole cycle of the chaperone-mediated protein folding is computationally challenging by using molecular dynamics.Therefore we focus on the SBD part of the Hsp70.Starting from the closed state prebound with the unfolded substrate hTRF1, we simulated the conformational switching of the SBD from the closed state to the open state,which corresponds to the nucleotide exchange step(Fig.1(a)).We then calculated the folding time of the substrate hTRF1 starting from the unfolded conformation bound at the open state of SBD with different chaperone–client interaction strengthsCHP.Figures 2(b) and 2(c)) show the mean first passage time (MFPT) of the substrate folding with two different criteria in assigning the successfully folded state: (i)theQvalue (i.e., the fraction of formed native contacts) is larger than 0.8 and the substrate protein is released from the chaperone; (ii) theQvalue is larger than 0.8 without considering whether the substrate is release or not.For comparisons,we also calculated the MFPT for substrate release.One can see that without considering the contribution of substrate release to the folding time, the MFPT of the substrate folding decreases with the increasing ofCHP,which is consistent with previous work.[21]In contrast,when considering the substrate release step,the MFPT decreases first and then increases with the further increasing ofCHP.Interestingly,the slowing down of the protein folding at higherCHPis not due to the waiting time of the substrate release,because the time of substrate release is much shorter even whenCHPis high for the parameter range studied in work, although the substrate release time increases withCHP.For example,at theCHPof 3.9,the folding time is 15.31×107MD steps, whereas the substrate release time is 1.07×107MD steps,which is 14 times shorter than the folding time.It is worth noting that the folding time with the criterion (i) cannot be directly calculated by the folding time with the criterion (ii) and the substrate release time because the whole folding process cannot be simply decomposed into folding and release steps,particularly at highCHPvalues.
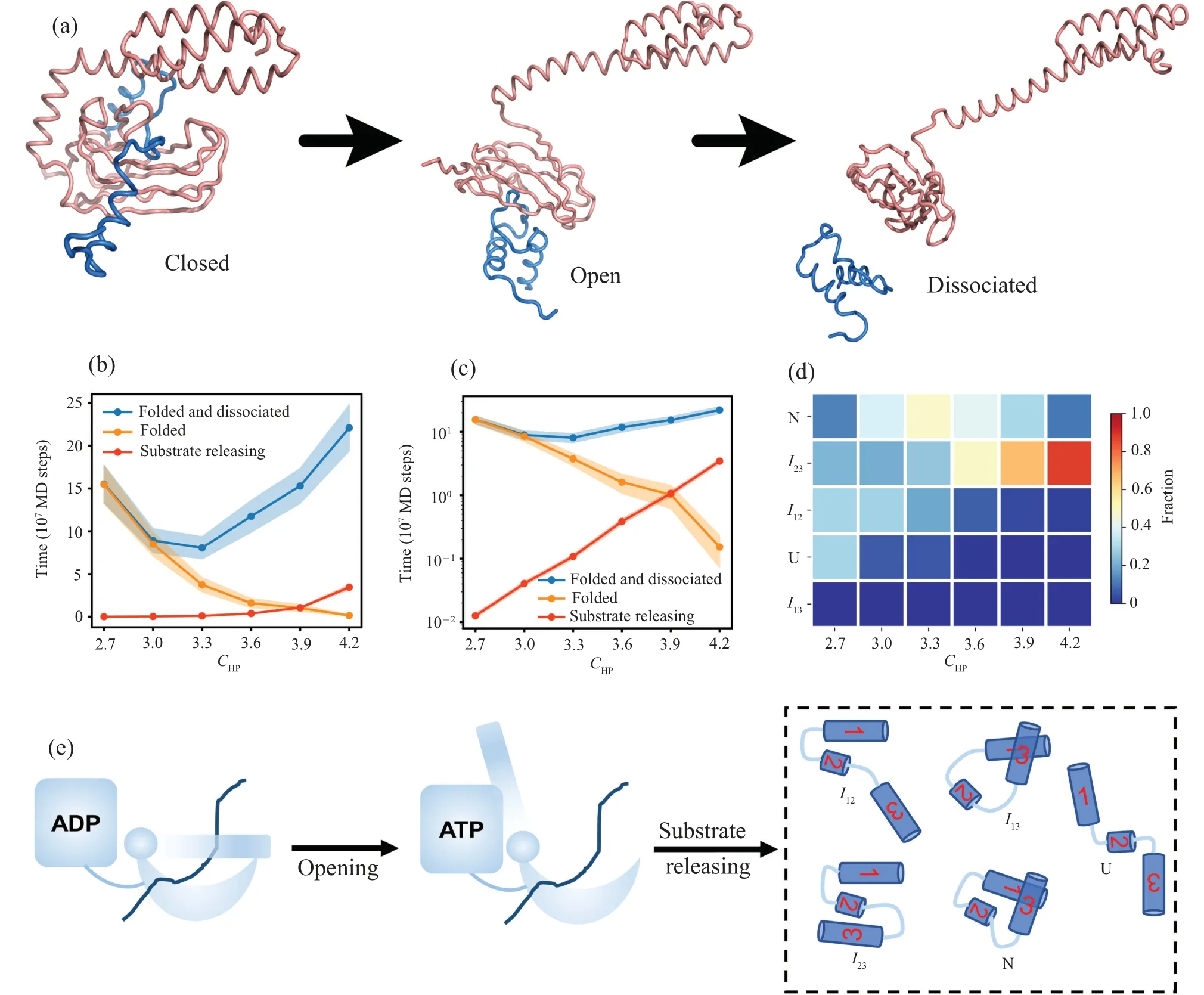
Fig.2.Hsp70 regulates the folding of the substrate protein hTRF1.(a) Representative snapshots at three different stages of the Hsp70-mediated hTRF1 folding.(b)The mean first passage time(MFPT)for the successful folding of the substrate, i.e., the substrate is well folded and released from the Hsp70(blue),as a function of chaperone–substrate interaction strength CHP.For comparison,we also plotted the MFPT of substrate folding without considering the substrate release step (orange) and the MFPT for the substrate release (red).The standard errors are shown by shaded areas.Panel (c) is the same as panel (b) but with the y axis being shown by logarithm scale.(d) Cartoon diagram showing the possible conformational states of hTRF1 right before the substrate releasing step.(e)Probabilities of different conformational states of hTRF1 right before the substrate releasing step at different chaperone–substrate interaction strength CHP.
During the folding of the substrate protein, it can involve five possible states, including the unfolded state (U),correctly folded state (N), on-pathway intermediateI12, onpathway intermediateI13, and misfolded stateI23.To understand the molecular mechanism of slowing down for protein folding when considering the substrate release,we calculated the probability of the events in which the substrate is released from each of the above five states (Fig.2(e)), and the results were shown in Fig.2(d).At theCHPvalue of 2.7, the substrate tends to release at the unfolded state U or on-pathway intermediate stateI12.With the increasing ofCHP, the probability of events releasing from the correctly folded state (N)increases and that from unfolded state decreases.For example, at theCHPof 3.3, the substrate tends to dissociate from the correctly folded N state with the probability of around 0.5, whereas the probability of releasing event from the unfolded state U becomes negligible.Such result suggests that the substrate release step and the protein folding are well coordinated at the aboveCHPvalue.This is easy to be understood because the hydrophobic patch of H2, which corresponds to the chaperone binding site,tends to be buried in the interior of the folded hTRF1, which therefore weakens the chaperone–client interactions and promotes the substrate release.With the further increasing of theCHPvalues,the substrate tends to dissociate from the misfolded stateI23.For example, at theCHPvalue of 4.2,the probability of substrate release from the misfolded stateI23becomes 0.875,whereas that from the native state becomes 0.1.These results suggest that the slowing down of correct folding with strong chaperone–client interactions is resulted from the improved probability of reentering into the misfolded stateI23before the substrate release step.
As shown in Figs.2(b)and 2(c),increasingCHPtends to decrease the MFPT to access the folded state of the substrate protein at the chaperone-bound state.The previous computational study revealed that the observed acceleration for accessing the native state is resulted from the destabilization of the frustrated pathway (involving the misfolded stateI23) by the chaperone–client interactions.[21]Here, the increased probability of the misfolded stateI23sampled right before the substrate release step with highCHPmay suggest that increasing the chaperone–client interactions also have the tendency of increasing the chance for the substrate protein to access the misfolded stateI23.
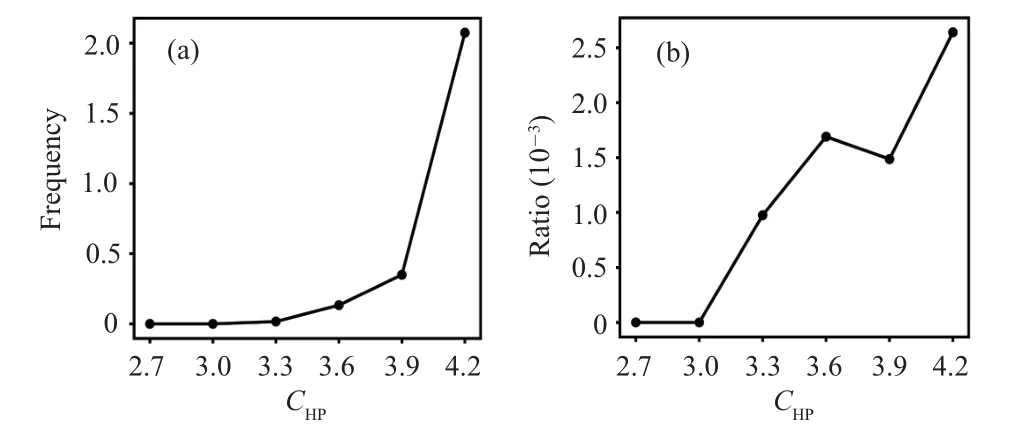
Figure 3 shows the frequency and relative ratio of transitions from all other states to the misfolded stateI23.One can see that before the substrate release step, increasingCHPfavors the transition from other states to the misfolded stateI23.Such result is somewhat counter-intuitive because it is clear that chaperone–client interactions tends to protect the substrate from being trapped in the misfolded state as shown in previous work.[21]
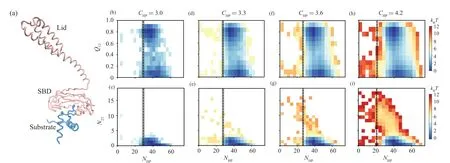
To further understand why increasing the chaperone–client interaction enhances the probability of accessing the misfolding state, we calculated the two-dimensional free energy landscape projected onto the reaction coordinateQ13–RSPandN23–RSPbased on the snapshots sampled before substrate release(Fig.4).HereQ13represents the fraction of formed native contacts between the H1 and H3.N23represents the number of contacts (mostly non-native contacts) formed between the H2 and H3.RSPrepresents the closest distance between the Hsp70 and client protein.One can see that at relatively weaker chaperone–client interaction strengths(e.g.,CHP=3.0 and 3.3), the misfolded stateI23is rarely sampled.The substrate dominantly adopts the unfolded conformation and the on pathway intermediatesI12/I13.With the further increasing ofCHP(e.g.,CHP=3.6 and 4.2),the probability of misfolded stateI23(the state with highN23value)increases.We also plotted the averageRSPvalue(dash line in Fig.4)calculated using the snapshot right before the substrate release step for each of the full folding events.One can see that with the increasing of the chaperone–substrate interaction strength,substrate has the chance to deviate from the chaperone by a relatively larger distance but still remains bound with the chaperone.The largerRSPvalue with highCHP,although still with minor population,may make the substrate have larger freedom to sample the misfolded state,as the hydrophobic patch is less protected by the chaperone.
We also observed the similar results from the twodimensional free energy landscape projected onto the reaction coordinatesQ13–NHPandN23–NHP(Fig.5).Here the reaction coordinateNHPrepresents the number of contacts between the Hsp70 and substrate.When theCHPvalue is low,a larger value ofNHPis needed to maintain the substrate being attached to the chaperone.However, with the increasing of theCHP, even small number of chaperone–client contacts can maintain the substrate attached.These results suggest that when the chaperone–client interaction strength is strong,even small number of inter-molecule contacts can maintain the substrate bound with the chaperone.Such sparsely contacted chaperone–client complex with longer chaperone–client distance releases the restraint of the substrate conformation by chaperone.The sampling of such transient chaperones–client complex with sparse inter-molecule contacts makes the client protein have chance to access the misfolded state even it is still bound with chaperone (Fig.3), which therefore tends to increase the probability that the substrate releases from the chaperone with the misfolded state as shown in Fig.2(e).

4.Conclusion
Chaperones are ubiquitous in both prokaryotic and eukaryotic cells, and play crucial role in maintaining the structural stability and functions of proteinsin vivo.[15]The functioning of chaperones involves multiple steps, such as the chaperone conformational change,cochaperone binding,substrate uptake and release,and ATP hydrolysis.[51,52]All these steps need to be delicately coordinated in order to achieve efficient and robust functions.Undoubtedly, the interaction strengths between all these participants need to be well optimized.Previous investigation revealed that the interactions between the chaperone and client protein at the open conformation may remodel the energy landscape and therefore direct the folding pathways to avoid misfolding.[21]In this work, we focused on the effect of the interaction strength between substrate and client at the open conformation.We found that there exists an optimal interaction strength between chaperone–client,at which the protein folding and release are well coordinated,and substrate tends to release from the chaperone at native state.When the interaction strength is low,increasing the strength favors the correct folding pathways and increases the folding rate (Fig.6).In contrast, when the interaction strength is high, increasing the strength slows down the folding.Such results provide new insights into the factors shaping up the high efficiency of the chaperone functioning,which can be valuable for the protein design.
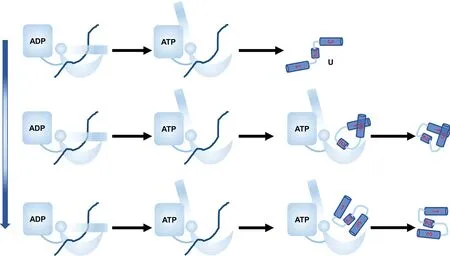
Fig.6.Cartoon diagram showing the effect of interaction strength CHP between chaperone and substrate on protein folding.When CHP is small,the substrate tends to release before the folding and the substrate tends to be trapped at misfolded state and folding is slow.At the optimal CHP, the substrate tends to release from the chaperone at the folded state,and increasing CHP speeds up substrate folding.At large CHP,the substrate tends to release from the chaperone at the misfolded state,and increasing CHP slows down the substrate folding.
Acknowledgements
The authors thank the kind help and insightful discussions with Wenfei Li and Wei Wang in Nanjing University.Project supported by the National Natural Science Foundation of China(Grant Nos.11305139 and 11974173)and the HPC Center of Nanjing University.

- Chinese Physics B的其它文章
- Optimal zero-crossing group selection method of the absolute gravimeter based on improved auto-regressive moving average model
- Deterministic remote preparation of multi-qubit equatorial states through dissipative channels
- Direct measurement of nonlocal quantum states without approximation
- Fast and perfect state transfer in superconducting circuit with tunable coupler
- A discrete Boltzmann model with symmetric velocity discretization for compressible flow
- Dynamic modelling and chaos control for a thin plate oscillator using Bubnov–Galerkin integral method