
儿童型低磷酸酶血症一例和基因突变分析
2023-11-27 11:14:54翟如玉姚琪米热古丽买买提
中国骨与关节杂志 2023年11期
翟如玉 姚琪 米热古丽·买买提
低磷酸酶血症 ( hypophosphatasia,HPP ) 是一种罕见的遗传性疾病,其特征是以骨及牙齿矿化缺陷,血清及骨碱性磷酸酶减低为主。该病的遗传方式为常染色体隐性或显性遗传,多数为常染色体隐性遗传,少数为显性遗传,发病率为 1 / 10 000,重型少见,轻型较常见[1]。由于家族内的可变表达和显性遗传方式的不完全外显,导致 HPP临床表现差异较大,即从严重无骨骼矿化的死产到没有骨骼症状的早期牙齿脱落,根据年龄可分为 6 种形式:围生期型 ( 致死性、良性 ),婴儿型,儿童型,成人型和牙型[2]。国外报道以围生期型和婴儿型为主[3],国内以儿童型和牙型为主,儿童型 HPP 的发病年龄在 6 月龄到 18 岁之间,症状严重程度不一[4]。笔者通过对 1 例儿童型 HPP患儿的临床、生化、影像以及基因检测结果的分析来探讨HPP 的诊断和发病机制。
病例资料
患儿,男,14 岁,为足月顺产,出生时四肢短小,母乳喂养,父母为近亲结婚,是表兄妹,兄弟姐妹 4 人,排行第三,家族成员无类似疾病史。1 岁 6 个月时发现脊柱向右侧侧凸,髋部扁宽,双小腿中段向前隆起,双侧肘关节外翻畸形,左手 2、3、4、5 指呈屈曲畸形,不能伸直,辗转就诊于多家医院未予以治疗。10 岁起患儿运动耐力逐渐减弱,由行走 500 米后出现劳累进展为行走 200 米后即觉劳累,遂就诊于北京积水潭医院,给予佩戴双下肢支具,佩戴 1~2 个月后患儿不适,未再佩戴,期间患儿运动耐力进行性下降。患儿于 11 岁时开始出现双小腿阵发性钝痛,行走时加重休息时可缓解,至 13 岁患儿出现行走20 米后即觉劳累,行走困难,遂再次就诊于北京积水潭医院,完善基因检测后诊断为“低磷酸酶症;脊柱侧凸;胫骨外翻”,未予以特殊治疗,期间可拄双拐行走至今。
体格检查:体重 42 kg,身高 132.2 cm,小于同龄儿童第三百分位数,-4.68 SD,头围 54 cm,大小正常无畸形,牙齿有松动脱落,具体表现为 16.26.36Ⅰ~Ⅱ° 松动,31.32.41.42 Ⅱ° 松动,46 缺失,双侧磨牙牙冠形态发育异常,牙列不齐。心肺腹 ( - ),肝脾未触及,全身皮肤检查无过度角化表现。鸡胸、肋骨外翻 ( 图 1a ),髋部扁宽,脊柱向右侧弯,活动度正常,四肢畸形:双小腿中段向前隆起 ( 图 1b、c ),右侧胫骨陈旧性骨折并畸形愈合。双侧肘关节外翻畸形,左手 2、3、4、5 指呈屈曲畸形 ( 图 1d ),不能伸直,非功能位活动受限。患儿神志清,精神佳,智力发育正常,平衡性可,情绪稳定,无异常行为,食欲好,大小便正常,体重正常增长,体力明显下降。
实验室检查:血常规检查结果均在正常参考值范围内,生化检查碱性磷酸酶 ( alkaline phosphatase,ALP ) 为31.80 U / L↓ ( 参考范围 38~126 U / L ),血清钙 2.32 mmol / L( 参考范围 2.10~2.55 mmol / L ),血清磷 2.25 mmol / L↑ ( 参考范围 0.81~1.45 mmol / L ),25 羟维生素 D 26.34 mmol / L↓( 参考范围 > 50 mmol / L ),血清骨钙素 113.10 ng / ml↑ ( 参考范围 14~46 ng / ml ),甲状旁腺激素 2.54 pmol / L ( 参考范围 1.6~6.9 pmol / L )。
影像学检查:X 线检查我院双下肢全长正位片图显示髋部扁宽,两侧骨盆对称,骨密度明显减低,骶髂关节间隙模糊 ( 图 1e );北京积水潭医院右侧肘关节正位片显示肘关节外翻畸形 ( 图 1f );胫 - 腓骨正侧位片显示双侧胫腓骨干弯曲:双下肢骨密度减低,右侧腓骨上段可疑透亮线,右侧胫骨中断骨皮质不连续,局部增生硬化并皮质增厚( 图 1g、h )。CT 检查我院全脊柱三维重建显示腰椎轻度 S 形侧凸畸形,骶尾椎曲度失常,椎体骨密度减低,骨皮质变薄,骨小梁稀疏 ( 图 1i );头颅三维重建未见异常 ( 图 1j );骨盆三维重建显示骨盆形态异常,呈“漏斗状”骨盆,骶尾椎曲度失常,骨密度减低,骨小梁稀疏 ( 图 1k )。
基因检测 ( 伦理批准号:20220308-15 ):为进一步明确诊断,在取得患儿家长知情同意并经医学伦理委员会审核后,采集患儿及家系成员外周血乙二胺四乙酸 ( ethylene diamine tetraacetic acid,EDTA ),由首都儿科研究所附属儿童医院对患儿进行全外显子组测序,通过目标区域捕获和高通量测序,并结合 Sanger 测序验证得出结果:患儿组织非特异性碱性磷酸酶 ( tissue non-specific alkaline phosphatase,TNSALP ) 基因存在一处纯合突变为 c.1555-1561 delinsGCTCAG ( 缺失 - 插入 ) 导致氨基酸改变 p.p519Afs*87( 移码突变-87 位后终止 ),突变分别来自于其父母,其妹及其姐此位点均无突变 ( 图 2 )。
讨 论
儿童型 HPP 通常表现为乳牙过早脱落,骨折,骨骼佝偻病样改变 ( 方颅,肋骨串珠样改变,膝外翻和膝内翻等畸形改变 ),骨骼疼痛和肌肉无力,严重者导致行走迟缓、蹒跚步态,部分患儿终身矮小[5]。乳牙过早脱落几乎是所有类型 HPP 共有的特征[6],若仅有牙齿症状,则为表型最轻的牙型 HPP。此患儿 1 岁时发病至今已有 13 年,除了有牙齿松动脱落外,还有胫骨陈旧性骨折,脊柱侧凸、长骨弯曲变形和外翻畸形,且近 5 年来已有不同程度的行走困难和步态异常,身材矮小等一系列特征符合儿童型 HPP 的表现。
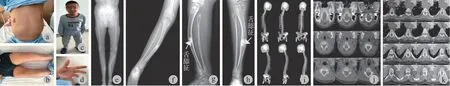
图1 a:鸡胸、肋骨外翻;b:胫骨向上隆起;c:骨骼畸形;d:左手 2、3、4、5 指呈屈曲畸形,不能伸直;e:双下肢全长正位 X 线片示髋部扁宽;f:右侧肘关节正位 X 线片示肘外翻畸形;g、h:右侧胫腓骨侧位和正位 X 线片示胫骨中段向上隆起,干骺端增宽,呈“舌舔征”( 箭头 );i:全脊柱三维重建示腰椎 S 形侧凸畸形;j:头颅三维重建未见异常;k:骨盆三维重建示“漏斗状”骨盆,骶尾椎曲度失常Fig.1 a: Chicken breast, ribs turned outward; b: The tibia bulged upwards; c: Skeletal deformity; d: The 2nd, 3rd, 4th, and 5th fingers of the left hand were flexed and could not be straightened; e: Both lower limbs were full-length and AP radiographs showed a broad hip; f: AP x-rays of the right elbow joint showed elbow valgus deformity. g-h. Lateral and AP radiographs of the lateral tibial fibula showed that the mid-tibia was raised upwards, and the metaphysis was widened, showing a “tongue-licking sign” ( arrow ); i: Three-dimensional reconstruction of the entire spine showed S-shaped scoliosis of the lumbar spine; j: There were no abnormalities in the three-dimensional reconstruction of the skull; k: Threedimensional reconstruction of the pelvis showed a “funnel-shaped” pelvis with an abnormal curvature of the sacrococcygeal vertebrae
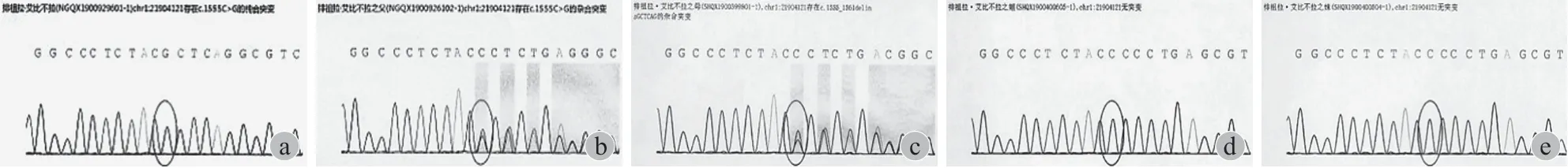
图2 患儿及家属全外显子组测序结果 a:患儿纯合突变;b:患儿父亲杂合突变;c:患儿母亲杂合突变;d:患儿姐姐无突变;e:患儿妹妹无突变Fig.2 Whole exome sequencing results of the child and family a: The child was homozygously mutant; b: The father of the child was homozygously mutant; c: The mother of the child was homozygously mutant; d: The child's elder sister had no mutation; e: The child’s younger sister had no mutation
骨源性 ALP 由成骨细胞产生,是儿童期 ALP 的主要来源,所以 ALP 活性降低是 HPP 的一个标志性指标,此患儿 ALP 指标降低是经过多次检验的结果,符合该特异性表现。国外曾有研究报道此疾病的严重程度与 ALP 活性高低相关,ALP 活性越低临床表现越严重[7],但此患儿ALP 并未显著降低临床表现却较为严重,这是因为 ALP的参考范围与年龄及性别相关,婴幼儿期和青春期时通常偏高,且女孩比男孩更早出现升高,所以不能仅仅以成人ALP 参考范围评估儿童[8],更提示诊断不能仅依靠生化指标,更需结合辅助检查和基因检测。
影像学检查在 HPP 的诊断中发挥着很大的作用,轻症者 X 线以佝偻病表现为主:如临时钙化带薄弱或消失,干骺端增宽,骨密度减低导致易弯曲骨折[9];重症者可表现为骨化不全,长骨干骺端呈锯齿状凹凸不平,骨化不均,骨干弯曲[10]。此患儿 X 线典型表现为四肢长骨弯曲,胫骨陈旧性骨折,干骺端增宽、呈“舌舔征”,骨骼畸形如鸡胸、肋骨外翻、髋部扁宽,左手 2、3、4、5 指呈屈曲畸形,符合 HPP 表现。HPP 患者出现骨折、佝偻病征象等骨矿化障碍表现通常被认为是由于 TNSALP 活性减弱或缺失,导致无机焦磷酸盐 ( pyrophosphoric acid,PPi )、磷酸吡哆醛 ( pyridoxal phosphate,PLP ) 和磷酸乙醇胺 ( phosphoethanolamine,PEA ) 的细胞外聚集和局部磷酸盐浓度降低[11]。
HPP 是由 ALPL 基因编码 TNSALP 过程中功能缺失使骨骼和牙齿矿化不足而导致的[2]。ALPL 基因位于1p36.12,约 50 kb 大小,共包含 12 个外显子[12],当 ALPL基因片段发生缺失、插入或者突变时,由于功能不足使TNSALP 的活性降低。PPi 在 TNSALP 的水解下生成无机磷,与钙结合形成的羟基磷灰石有促进骨骼矿化的作用,当 ALP 下降时不仅影响羟基磷灰石的生成,也使 PPi 堆积在骨骼中。PPi 有抑制骨骼矿化的作用,堆积时造成骨骼矿化障碍[13]。基因突变存在地域差异,加拿大马尼托巴省的发病率最高,尤其以围生期型 HPP 发病为主[7]。基因突变的类型目前日本以 12 号外显子上的 c.1559delT最为常见,欧美国家分别以 6 号 c.571G>A 和 10 号外显子c.1133A>T 为主要变异[14],我国以第 5 外显子区 c.407G>A变异最为常见且类型以错义突变最为常见,其次是插入与缺失[4],插入与缺失常导致移码突变和整码突变,患儿基因突变检测结果显示 ALPL 基因处存在 12 号外显子上的c.1555-1561delinsGCTCAG ( 缺失 - 插入 ) 印证了以上诊断,此突变在国内外研究中均未见报道,推测为一种新的突变。由于 12 号外显子上的 c.1555-1561delinsGCTCAG ( 缺失 - 插入 ) 移码突变导致的翻译提前终止,使多肽链的氨基酸顺序发生改变从而导致蛋白质性质发生改变,截短蛋白 ( p.P519Afs*87 ) 丧失了发挥酶活性和骨矿化作用的重要区域;突变分别来自于父母,父母均为杂合突变携带者表型正常,患儿为纯合突变,符合常染色体隐性遗传方式。目前已报道的 HPP 如围生期型、婴儿型通常都为复合杂合子突变,很少见纯合突变[15]。HPP 需要与常见的易导致骨骼畸形的其它疾病相鉴别,如维生素 D 依赖性佝偻病、成骨发育不全等;佝偻病的 ALP 水平常升高并伴低磷或低钙血症,成骨发育不全以反复骨折、低骨密度为主要表现,血钙、血磷和 ALP 水平正常,均可通过生化检查初步鉴别。
HPP 通常以临床对症治疗为主,特异性酶替代疗法和重组人甲状旁腺素 1-34 ( 特立帕肽 ) 为广大学者所推崇,但特立帕肽可诱导儿童生长板内发生骨肉瘤,不推荐用于儿童患者[16],而酶替代疗法 ( enzyme replacement therapy,ERT ) 已经成为日本、欧美等国家目前最佳的治疗方法,但是国内还并未引进此药物[11]。ERT 在儿童期 HPP 的治疗中不仅能够减少骨骼肌肉的疼痛,还可以改善肌力运动以及生长发育[17],若能在患儿年龄较小时早发现早诊断早治疗,酶替代治疗是可以改善最终身高和骨骼畸形的。对症治疗如非甾体类抗炎药可用于骨痛、骨关节炎、骨软化的治疗[18]。此患儿经过北京协和医院与上海交通大学医学院附属新华医院的远程会诊,建议尽可能进行酶替代疗法,随后患儿去美国进行 ERT,目前仍在长期随访中。关于预后,儿童 HPP 被认为是一种稳定的疾病。然而,牙齿和骨骼疾病以及关节受累会在成年期重新出现,因此,在儿童期或青春期诊断的 HPP 不应该被视为良性疾病,应当定期监测[19]。
综上所述,ALPL 基因中的 c.1555-1561delinsGCTCAG突变是该病的致病基因,并且是尚未报道过的新的突变基因,有助于更进一步探讨 HPP 的发病机制,从而更准确地诊断与治疗。同时,在诊断方面应更加关注骨关节的病变及骨骼畸形的原因与佝偻病、成骨不全等疾病相鉴别,在治疗方面应及早引进国外的酶替代治疗药物,且患儿需遵循医嘱长期随访和定期监测避免预后不良。
猜你喜欢
建材发展导向(2022年24期)2022-12-22 07:44:36
电子科技大学学报(2022年5期)2022-10-29 01:57:52
环境卫生工程(2021年4期)2021-10-13 06:52:26
中国生殖健康(2020年4期)2021-01-18 02:58:10
软件(2020年3期)2020-04-20 00:56:34
中国生殖健康(2018年4期)2018-11-06 07:12:16
光学精密工程(2016年6期)2016-11-07 09:07:56
腹腔镜外科杂志(2016年12期)2016-06-01 12:10:09
中国医疗美容(2015年1期)2015-07-12 10:05:59
湖北农业科学(2014年11期)2014-09-10 18:06:07
