
USP18介导的蛋白质去ISG化及其在结核病等传染病中的作用
2023-11-24 07:17张其奥王子路李佩波谢建平
遗传 2023年11期
张其奥,王子路,李佩波,谢建平,
综 述
USP18介导的蛋白质去ISG化及其在结核病等传染病中的作用
张其奥1,王子路1,李佩波2,谢建平1,2
1. 西南大学生命科学学院现代生物医药研究所,重庆 400715 2. 重庆市公共卫生医疗救治中心,重庆 400036
干扰素诱导基因15 (interferon-stimulated gene 15,)的表达受Ⅰ型干扰素诱导,该基因编码的蛋白ISG15可以分别通过E1、E2和E3酶的作用共价修饰靶蛋白,此过程被称为ISG化(ISGylation)。宿主蛋白的ISG化广泛参与天然免疫例如宿主的抗病毒过程。泛素特异性蛋白酶18 (ubiquitin-specific protease 18,USP18)作为一种去泛素化酶(deubiquitinase,DUB)可以去除靶蛋白偶联的ISG15,并通过抑制Ⅰ型干扰素信号通路来抑制宿主的免疫应答。ISG15介导的ISG化和USP18介导的去ISG化(deISGylation)建立的动态平衡对结核病的发生、发展和转归有重要影响。此外,同ISG15一样,USP18也广泛参与病毒感染和宿主细胞抗病毒反应,多种先天性免疫疾病和免疫信号通路都受到USP18的调节。本文综述了ISG15和USP18相关的研究进展,重点介绍了ISG15介导的ISGylation和USP18介导的去ISG化在结核病及其他重要疾病中的调控作用,以期为靶向宿主蛋白的结核病等重要疾病防治提供新的策略。
USP18;ISG15;结核病;干扰素
泛素特异性蛋白酶18 (ubiquitin-specific protease 18,USP18)和干扰素诱导基因15 (interferon- stimulated gene 15,)介导的ISG化修饰是机体参与抗病原微生物感染的重要组分。越来越多的研究发现,靶向ISG15和USP18及其底物的蛋白质翻译后修饰有望开发成为新型抗感染治疗的策略之一。USP18是一个大小约43 kDa的特异性蛋白酶,ISG15和USP18均可被干扰素(interferon,IFN)诱导。通过比较人类()、家鼠()、家牛()、野猪()、罗伯罗夫斯基仓鼠()、绿头鸭()等物种中USP18的氨基酸序列,发现不同物种中的USP18具有高度的同源性。USP18在不同物种中都含有高度保守的序列,该保守序列包括半胱氨酸残基和组氨酸残基位于USP18的活性中心,这也是USP家族蛋白酶所特有的结构(图1)。ISG15能够参与并激活多种信号通路,发挥抗病毒免疫的功能,与之相反,USP18会限制NF-κB、JNK和NFAT等通路的激活,负调节炎症反应,调节T淋巴细胞和辅助性T细胞的活化。USP18在Ⅰ型IFN中的作用下依赖于α/β干扰素受体2 (interferon alpha/beta receptor 2,IFNAR2),并负调控Ⅰ型IFN信号通路。USP18可以切割ISG15与靶蛋白之间的异肽键,还能在结合后立即切割ISG15的LRLRGG序列,在由ISG15前体加工为成熟的ISG15过程中发挥重要作用。鉴于ISG15和USP18在介导宿主蛋白泛素化和去泛素化以及免疫应答中的重要作用,本文主要关注ISG15和USP18及其突变体在结核分枝杆菌(,Mtb)等感染免疫应答中的作用及其作为治疗靶点的潜力。
1 ISG15特异性蛋白酶
蛋白去泛素化是由一组去泛素酶(deubiquitinase,DUB)介导的泛素化的反向过程。DUB家族有90多个成员[1]。泛素特异性蛋白酶USP18和USP20属于DUBs的USP亚家族,并介导靶蛋白的去泛素化。USP20靶向多种蛋白质底物,包括HIF1α、β-肾上腺素能受体、TNF受体关联因子6(TNF receptor associated factor 6,TRAF6)和Claspin,通过去泛素化调节Toll样受体4(toll-like receptor 4,TLR4)信号转导和DNA损伤修复[2-6]。USP18最初被鉴定为DUB,后来被发现也具有去ISG化酶活性[7,8],因为小鼠中缺失或非活性突变体usp18会导致高水平的ISG化[9,10]。此外,USP18在ISG15前体加工产生成熟的ISG15分子过程中也发挥作用[11],但在缺陷小鼠中,ISG15前体也能被加工成其成熟形式[12],重组ISG15前体可以被II型肺泡上皮细胞A549的一种大小100 kDa的酶正确加工,且该酶是酵母泛素特异性肽酶1(ubiquitin-specific protease,USP1)同源物,活性不受I型IFN刺激的影响[12]。这提示除USP18之外,还有其他ISG15特异性蛋白酶。一些E2和E3酶在ISG15化和泛素化过程中的功能具有重叠,也意味着存在可以作为ISG15特异性蛋白酶的多功能DUB[13],包括USP2、USP5、USP13和USP14在内的几种DUB都被认为是ISG15特异性蛋白酶的候选物[8]。但是,小鼠中基因的缺失会导致组织中ISG15结合物大量增加,而不会影响泛素结合物的水平,这表明USP18是ISG15特异性蛋白酶,可将ISG15从靶蛋白上去除(图2)。

图1 USP18蛋白在不同物种中保守
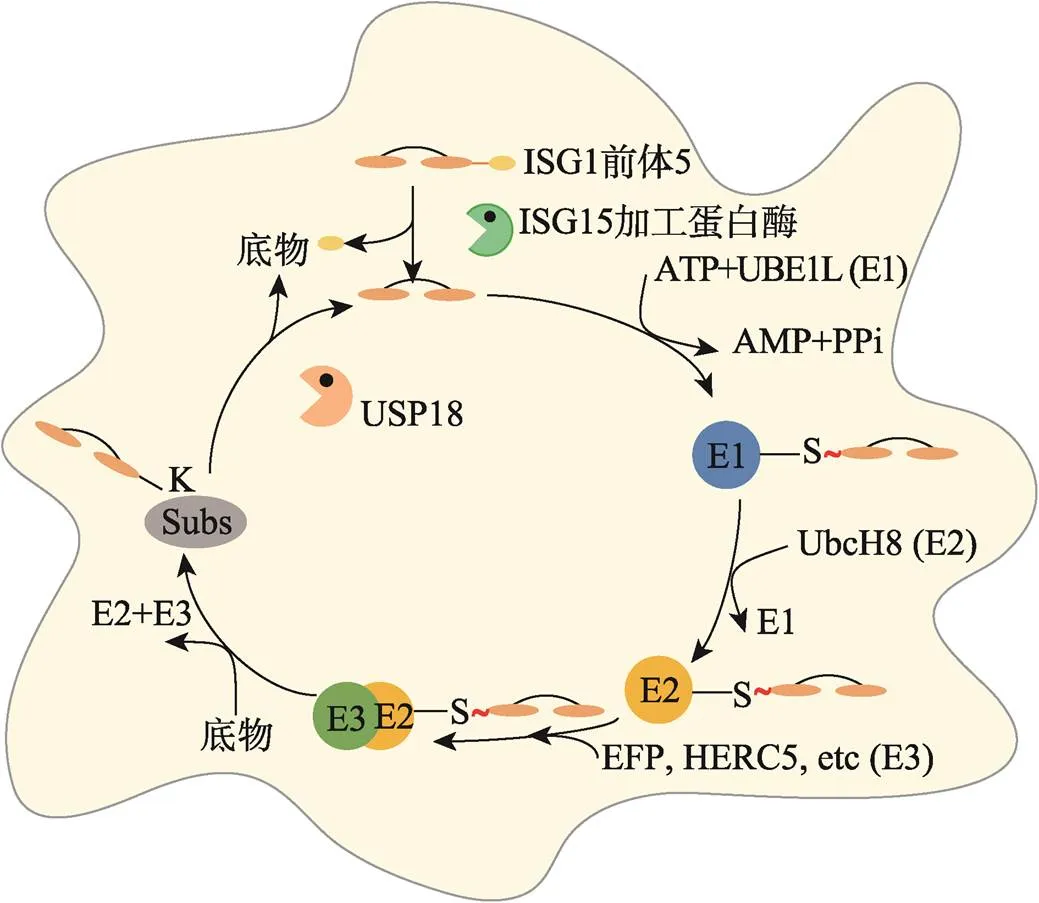
图2 ISGylation过程
ISG15前体经ISG15加工酶变成ISG15,分别与E1、E2、E3泛素酶结合,将ISG15结合在靶蛋白上,USP18能够从靶蛋白上去除ISG15,发挥去ISG化功能。
USP18的表达主要受I型IFN诱导,而这种诱导需要通过JAK/STAT信号通路的作用[8,14]。干扰素β (interferon-beta,IFN-β)比干扰素α (interferon-alpha,IFN-α)和双链RNA (double strand RNA,dsRNA)诱导USP18的作用更强,但干扰素γ (interferon-gamma,IFN-γ)几乎没有诱导作用[13]。USP18也可被脂多糖(lipopolysaccharide,LPS)诱导。干扰素调节因子2 (interferon regulatory factor 2,IRF2)和干扰素调节因子3 (interferon regulatory factor 3,IRF3)都是LPS应答所必需的,LPS通过IRF3上调USP18,而IRF2可将其降至基础水平。在各种造血细胞系中,单核细胞和巨噬细胞系能够高水平表达USP18[15]。Skp2 (S-phase kinase associated protein 2)是S期激酶相关蛋白,属于F-box蛋白家族,在蛋白泛素化降解过程中可作为Skp1-Cul1-F-box(SCF)蛋白复合物的重要成分识别底物蛋白,通过降解细胞周期调节蛋白而调控细胞周期。USP18是Skp2的底物,Skp2促进USP18的泛素化和随后的蛋白酶体降解。这表明SCF-Skp2可能通过控制USP18的稳定性,调节I型IFN信号转导[16]。然而,有大量基因例如急性髓系白血病(acute myeloid leukemia,AML)融合基因诱导USP18上调的机制尚待研究。
2 USP18负调节先天免疫反应
ISG15和USP18在宿主对病毒感染的反应中起着重要作用。在I型IFN处理后,与野生型细胞相比,usp18细胞对B型流感病毒感染有更强的抵抗力,而病毒在缺失的usp18细胞中可完全恢复复制能力[10]。除了其酶功能外,USP18还负调控I型IFN信号转导,与Janus激酶(janus kinase,JAK)竞争结合IFNAR2[17]。因此,与野生型细胞相比,用I型IFN处理细胞导致信号转导转录激活因子(signal transducers and activators of transcription 1,STAT1)磷酸化增加且持续时间延长,增强ISG化,并促进细胞凋亡[11]。这与抑制USP18可增强HepG2.2.15细胞中IFN-α的抗乙型肝炎病毒(hepatitis B virus,HBV)活性这一结论相吻合。接下来文章将重点阐述USP18作为先天免疫反应负调节剂所参与的经典信号通路及机制(图3)。
模式识别受体如RIG-I在先天免疫细胞中表达,招募接头分子IPS-1,进而触发MAVS-TBK1-IRF3通路等信号通路的激活,ISG15能够靶向并参与修饰一系列重要的抗病毒蛋白,如STAT1、IRF3、RIG-I等,病毒感染过程中ISG15介导的ISGylation以及与底物的结合能够使宿主发挥抗病毒反应,同时USP18将负调控某些抗病毒蛋白的表达。
2.1 USP18与I型IFN信号通路
与ISG15一样,USP18在宿主先天免疫中发挥重要作用。这种作用分别通过ISG15蛋白酶依赖性和非依赖性方式介导。USP18由IFN、LPS和病毒感染诱导,并可调节I型IFN应答[18]。小鼠中基因的缺失导致IFN超敏反应[11]。小鼠对多种病毒(包括淋巴细胞脉络丛脑膜炎病毒(virus,LCMV)、水疱性口炎病毒(virus,VSV)和辛德比斯病毒(virus,SINV))感染引起的细胞病变效应也具有更强的抵抗力[11]。缺陷小鼠在脑内接种LCMV和VSV后,未见致命性淋巴细胞脉络丛脑膜炎和脊髓脑炎[19]。此外,LCMV感染小鼠后,LCMV复制被严重抑制,大脑中ISG15化水平增加,小鼠的存活率大大提高。的小鼠感染LCMV后第11天都没有死亡或出现临床症状,而所有LCMV感染的野生型小鼠在第7天死亡。这些发现表明,缺失不利于LCMV复制[20]。
USP18可负调节针对病毒感染的先天免疫应答。小鼠胚胎成纤维(mouse embryonic fibroblast,MEF)细胞I型IFN介导的免疫反应增强,对VSV和SINV感染产生抗性,使STAT1信号失调。然而,在/isg15或/ube1l双敲除小鼠中,上述表型依旧没有改变,这表明与ISG15的蛋白质修饰无关。因此,USP18也受到ISG15外其他蛋白的调控,具有异肽酶活性。这些功能未必需要ISG15蛋白酶活性来介导,否则usp18小鼠的表型将被逆转,或至少受到ISG15或其E1激活酶UBE1L的敲降的影响,但事实上两者都没有影响[20,21]。USP18可能通过与参与免疫调节的蛋白质直接相互作用来影响免疫功能。比如USP18与IFN受体(IFNAR2)结合,并通过破坏IFNAR2-JAK结合来阻断IFN信号传递[18]。这些数据表明,USP18可以结合并调节蛋白的活性,而不依赖于其ISG15蛋白酶功能。
usp18细胞对I型IFN敏感,这与JAK/STAT信号通路的增强与延长有关。USP18除了具有催化活性,还可通过与IFNAR2 (I型IFN受体的亚基)之间的直接相互作用实现对I型IFN信号通路的负调控。IFNAR2与USP18的结合干扰JAK蛋白与受体之间的相互作用,导致下游磷酸化级联和其他信号的抑制。此外,在usp18细胞中通过siRNA敲降,其STAT2磷酸化与在usp18细胞几乎一致。这表明USP18抑制I型IFN信号通路不需要其去ISG化活性。这进一步证实了usp18表型的改变不受缺失的影响[22]。
2.2 USP18与STING信号通路
cGAS-STING_(cyclic GMP-AMP synthase- stimulator of interferon response CGAMP interactor,cGAS-STING )通路广泛参与细菌感染导致的疾病,例如结核病和败血症[23]。STING招募TANK结合激酶1(TANK-binding kinase 1,TBK1)和IRF3。TBK1首先被磷酸化,然后磷酸化的TBK1再磷酸化IRF3,IRF3进一步转移到细胞核中,诱导IFN-I和许多其他炎性细胞因子的表达。线粒体代谢酶ACOD1 (aconitate decarboxylase 1;也称为immune-responsive gene 1 protein homolog,IRG1)在巨噬细胞激活后被迅速诱导,催化顺乌头酸脱羧并产生高浓度衣康酸(itaconate,ITA),Mtb诱导的IRG1反应高度依赖于细菌ESX-1分泌系统以及宿主STING和I型IFN受体信号转导[24],USP18参与STING的调节[25]。
cGAS对于宿主防御细胞中的DNA病毒至关重要。USP18是STING的相互作用蛋白,作用于蛋白N端跨膜结构域[26]。导致单纯疱疹病毒1 (virus 1,HSV-1)或细胞质DNA受损,从而触发IRF3和核转录因子κB (nuclear factor kappa-B,NF-κB)的激活,随后在IFNAR1存在或不存在的情况下诱导产生I型IFN和促炎细胞因子。USP18与STING相互作用并招募USP20,USP20介导STING的K48-多聚泛素链的去泛素化。的缺失或的敲降导致的泛素化增强和STING的降解,增强了HSV-1病毒的复制,使usp18小鼠更易感染HSV-1,说明USP18是病毒感染后诱导I型IFN和促炎细胞因子所必需的,并且在DNA病毒触发的信号转导中起主要作用。USP18也被证明能直接充当DUB,并去除TAK1(transforming growth factor-beta-activated kinase 1,TAK1)的K63连接的多泛素链[27]。与此同时,USP18也能够独立于IFN-I活性或其DUB活性,进而介导抗病毒信号转导[26]。以上说明USP18通过依赖或独立于IFN和其酶活性的方式与不同的信号通路相互作用。
2.3 USP18与MAVS信号通路
线粒体抗病毒信号蛋白(mitochondrial antiviral signaling protein,MAVS)是一种线粒体锚定蛋白[28],在RIG-I样受体(rig-I-like receptors,RLRs)激活后指导对RNA病毒感染的先天免疫反应,最终通过IRF3触发I型IFN诱导并通过NF-κB引起炎症反应,分泌细胞因子[29-31]。许多E3泛素连接酶参与MAVs信号转导,例如TRAF6[32]、ITCH和RNF11[29],但对结核病中参与MAVS信号通路的USP18分子研究甚少。
USP18可以显著促进MAVS的多泛素化。病毒感染后增强USP18和MAVS之间的相互作用,这表明USP18可能调节MAVS活性。此外,USP18促进K63相关的多泛素化和随后的MAVS聚集。因此,巨噬细胞或成纤维细胞中导致病毒感染后I型IFN反应受损。小鼠更容易感染病毒。USP18对MAVS的影响与其酶活性无关,但取决于三部分基序蛋白31 (tripartite motif containing 31,TRIM31)。在病毒感染后,USP18对于TRIM31从细胞质转移到线粒体以及TRIM31和MAVS之间的相互作用至关重要[33]。
2.4 USP18与JAK/STAT信号通路
当宿主感染病原体后,人体刺激自身免疫系统抑制病原体的入侵和复制。其中抵抗病毒最有效的方法是产生IFN,它可以刺激人体免疫细胞(如巨噬细胞和自然杀伤细胞),增强宿主的防御能力。IFNAR1和IFNAR2被激活后,它们分别与下游酪氨酸激酶2 (tyrosine kinase 2,TYK2)和JAK1结合,激活的TYK2和JAK1反过来磷酸化IFNAR2相关的STAT2和STAT1,从而形成DNA结合的STAT1- STAT2-IRF9三元复合物(ISGF3)。STAT2是I型IFN信号转导的特异性效应物[34],能够与USP18直接互作[35],IFN-I诱导的USP18与JAK1竞争结合IFNAR2,IFNAR2也受到STAT2的辅助,并负调控IFN-I和JAK/STAT途径。因此除了是IFN信号转导的关键效应物外,STAT2对于USP18介导的JAK-STAT信号转导抑制至关重要。
3 USP18先天缺失与相关疾病
基因的缺失使细胞内的IFN水平失调,一方面可能会导致个体出现先天致死,但另一方面也能够增强癌症患者或病毒感染患者的免疫应答,促进癌症治疗和病毒清除。和缺失的致病机制在于IFN-I信号通路的负反馈控制受损。作为对IFN-α刺激的响应,USP18被上调表达,并负责抑制IFN-I途径,以减轻IFN-α炎症水平[36,37]。细胞内ISG15则与这种负反馈调节器的调控保持动态平衡[38]。因此,当缺失时,ISG的高水平和相关表型是由于IFN-I反应的失调,而不是IFN-I诱导的失调,类似I型IFN病中的情况。缺失的个体易患孟德尔遗传易感性的分枝杆菌病(mendelian susceptibility to mycobacterial disease,MSMD),这是因为缺失会触发T细胞和自然杀伤细胞(natural killer cell,NK)产生过量的IFN-γ[39]。小鼠更容易感染Mtb,肺和脾脏中的细菌数量增加,炎性细胞因子升高,肺部病变更严重。相比之下,常染色体隐性遗传(autosome recessive,AR)的完全缺失,大大增强IFN-I信号,在缺乏适当治疗的情况下,个体出生时是致命的,并且与感染易感性无关[40,41]。
重要的是,与缺陷患者不同,usp18个体在体外全血测定中IFN-γ/IL-12轴没有缺陷。然而,持续的IFN-I反应,特别是在髓系细胞中,降低了其IL-12和IL-23的产生[42]。usp18导致患者对分枝杆菌易感,无法有效抑制I型IFN通路,这不是因为该基因产物本质上有助于控制分枝杆菌感染,而是因为该等位基因不能控制相关的IFN-I炎症。这表明缺陷个体的BCG相关疾病可能是由IFN-I失调导致的。除了去ISG化外,USP18从T细胞受体(T cell receptor,TCR)或TNF信号转导下游的TAK1/TAB1复合物中去除K63连接的多泛素链,并限制NF-κB和NFAT的过度激活[27,43],从遗传学角度提示免疫过度活跃和免疫缺陷共存[44]。因此,USP18在不同的信号通路中发挥去ISG化、去泛素化和非酶活性。USP18是否以及如何参与病毒感染触发的信号转导还需要进一步探索。
ISG15/USP18通路调节细胞功能,对宿主感染慢性病毒感染(如丙型肝炎病毒(hepatitis C virus,HCV))的先天免疫反应至关重要[45]。研究表明ISG15/USP18通路在HCV感染的肝组织中发生改变,在调节HCV感染和对IFN治疗的耐药性中起重要作用。USP18的异肽酶非依赖性作用也参与HBV的复制[17]。细胞显示出对I型IFN诱导ISG应答增强,这表明的缺失导致免疫应答增强。同时,与小鼠相比,小鼠中HBV DNA的稳定性显著降低。因此,调节USP18表达水平有望治疗病毒感染,特别是对I型IFN信号敏感的病毒。除了抵抗HBV外,缺陷还增加了白血病融合蛋白(breakpoint cluster region-abelson,BCR- ABL)致癌转化的抗性。BCR-ABL是由BCR的N-末端部分与ABL酪氨酸激酶结合而成。这说明白血病的抗性在很大程度上取决于I型IFN信号通路的激活。通过IFNAR缺失阻断I型IFN信号通路可逆转细胞对白血病的初始抗性[46]。因此,抑制USP18对I型IFN信号通路的负调控可能会增强针对癌症的先天免疫应答[47]。
小鼠呈现骨质疏松症,这与缺陷破骨细胞前体中破骨细胞分化因子(receptor activator of nuclear factor kappa-b ligand,RANKL)信号增强有关。在RANKL介导的缺陷型小鼠骨髓源巨噬细胞(bone marrow-derived macrophage,BMM)向破骨细胞分化过程中,一些细胞因子表达水平的异常增加可能触发了破骨细胞高强度分化。尽管已知I型IFN会限制破骨细胞分化,但敲除小鼠中I型IFN反应的过度激活会导致小鼠骨质减少[48]。AP-002(基于镓的抗癌口服化合物,用于骨转移癌症患者的新型临床治疗)可显著逆转RANKL诱导的基因表达[49]。在甲状旁腺功能减退状态下,甲状旁腺激素(parathyroid hormone,PTH)低于正常值会导致高循环磷酸盐,碳酸酐酶II (Carbonic anhydrase II,CA-II)和USP18表达降低。CA-II在尾状组织中的表达减少以及随后钙吸收作用的丧失将使成骨分子的平衡向钙化前状态倾斜,导致基底节钙化[50]。
4 结语与展望
蛋白质翻译后修饰是蛋白质充分发挥生物活性的重要过程之一,也是疾病诊疗标志物、药物靶点等的重要来源。蛋白质泛素化修饰是感染免疫的重点研究对象之一,去泛素酶USP18是维持细胞中被ISG15共价修饰蛋白质的动态平衡和功能的关键,在DNA/RNA病毒感染期间调节病毒复制、聚集以及对宿主的易感性。ISG15作为一种类泛素蛋白也在抗病毒感染、宿主免疫应答、细胞周期等方面发挥重要作用。细胞高水平表达ISG15,增强上百种ISG相关基因表达并提高了对IFN的敏感性。总之,HBV等病原可以逃避宿主模式识别受体的识别、抑制下游信号转导,调控宿主USP18等去泛素化酶活性而干扰IFN信号转导,最后逃避免疫清除。因此针对DUBs尤其是USP18在患者中的异常表达及其调控因子研发新的靶向药物,将有助于开发新的抗感染工具。
[1] Reyes-Turcu FE, Ventii KH, Wilkinson KD. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes., 2009, 78: 363–397.
[2] Li ZB, Wang DK, Messing EM, Wu G. VHL protein- interacting deubiquitinating enzyme 2 deubiquitinates and stabilizes HIF-1alpha., 2005, 6(4): 373–378.
[3] Berthouze M, Venkataramanan V, Li Y, Shenoy SK. The deubiquitinases USP33 and USP20 coordinate beta2 adrenergic receptor recycling and resensitization., 2009, 28(12): 1684–1696.
[4] Yasunaga J, Lin FC, Lu XB, Jeang KT. Ubiquitin-specific peptidase 20 targets TRAF6 and human T cell leukemia virus type 1 tax to negatively regulate NF-kappaB signaling., 2011, 85(13): 6212–6219.
[5] Yuan J, Luo KT, Deng M, Li YH, Yin P, Gao BW, Fang Y, Wu PQ, Liu TZ, Lou ZK. HERC2-USP20 axis regulates DNA damage checkpoint through Claspin., 2014, 42(21): 13110–13121.
[6] Zhu M, Zhao HC, Liao J, Xu XZ. HERC2/USP20 coordinates CHK1 activation by modulating CLASPIN stability., 2014, 42(21): 13074–13081.
[7] Malakhov MP, Malakhova OA, Kim KI, Ritchie KJ, Zhang DE. UBP43 (USP18) specifically removes ISG15 from conjugated proteins., 2002, 277(12): 9976–9981.
[8] Schwer H, Liu LQ, Zhou L, Little MT, Pan Z, Hetherington CJ, Zhang DE. Cloning and characterization of a novel human ubiquitin-specific protease, a homologue of murine UBP43 (Usp18)., 2000, 65(1): 44–52.
[9] Ritchie KJ, Malakhov MP, Hetherington CJ, Zhou LM, Little MT, Malakhova OA, Sipe JC, Orkin SH, Zhang DE. Dysregulation of protein modification by ISG15 results in brain cell injury., 2002, 16(17): 2207–2212.
[10] Ketscher L, Hannß R, Morales DJ, Basters A, Guerra S, Goldmann T, Hausmann A, Prinz M, Naumann R, Pekosz A, Utermöhlen O, Lenschow DJ, Knobeloch KP. Selective inactivation of USP18 isopeptidase activity in vivo enhances ISG15 conjugation and viral resistance., 2015, 112(5): 1577–1582.
[11] Ritchie KJ, Hahn CS, Kim KI, Yan M, Rosario D, Li L, de la Torre JC, Zhang DE. Role of ISG15 protease UBP43 (USP18) in innate immunity to viral infection., 2004, 10(12): 1374–1378.
[12] Potter JL, Narasimhan J, Mende-Mueller L, Haas AL. Precursor processing of pro-ISG15/UCRP, an interferon- beta-induced ubiquitin-like protein., 1999, 274(35): 25061–25068.
[13] Li XL, Blackford JA, Judge CS, Liu M, Xiao W, Kalvakolanu DV, Hassel BA. RNase-L-dependent destabilization of interferon-induced mRNAs. A role for the 2-5A system in attenuation of the interferon response., 2000, 275(12): 8880–8888.
[14] Malakhova OA, Yan M, Malakhov MP, Yuan YZ, Ritchie KJ, Kim KI, Peterson LF, Shuai K, Zhang DE. Protein ISGylation modulates the JAK-STAT signaling pathway ., 2003, 17(4): 455–460.
[15] Malakhova O, Malakhov M, Hetherington C, Zhang DE. Lipopolysaccharide activates the expression of ISG15-specific protease UBP43 via interferon regulatory factor 3., 2002, 277(17): 14703–14711.
[16] Tokarz S, Berset C, La Rue J, Friedman K, Nakayama KI, Nakayama K, Zhang DE, Lanker S. The ISG15 isopeptidase UBP43 is regulated by proteolysis via the SCFSkp2 ubiquitin ligase., 2004, 279(45): 46424–46430.
[17] Li L, Lei QS, Zhang SJ, Kong LN, Qin B. Suppression of USP18 potentiates the anti-HBV activity of interferon alpha in HepG2.2.15 cells via JAK/STAT signaling., 2016, 11(5): e0156496.
[18] Malakhova OA, Kim KI, Luo JK, Zou WG, Kumar KGS, Fuchs SY, Shuai K, Zhang DE. UBP43 is a novel regulator of interferon signaling independent of its ISG15 isopeptidase activity., 2006, 25(11): 2358–2367.
[19] Knobeloch KP, Utermöhlen O, Kisser A, Prinz M, Horak I. Reexamination of the role of ubiquitin-like modifier ISG15 in the phenotype of UBP43-deficient mice., 2005, 25(24): 11030–11034.
[20] Osiak A, Utermöhlen O, Niendorf S, Horak I, Knobeloch KP. ISG15, an interferon-stimulated ubiquitin-like protein, is not essential for STAT1 signaling and responses against vesicular stomatitis and lymphocytic choriomeningitis virus., 2005, 25(15): 6338–6345.
[21] Kim KI, Yan M, Malakhova O, Luo JK, Shen MF, Zou WG, de la Torre JC, Zhang DE. Ube1L and protein ISGylation are not essential for alpha/beta interferon signaling., 2006, 26(2): 472–479.
[22] Arimoto KI, Löchte S, Stoner SA, Burkart C, Zhang Y, Miyauchi S, Wilmes S, Fan JB, Heinisch JJ, Li Z, Yan M, Pellegrini S, Colland F, Piehler J, Zhang DE. STAT2 is an essential adaptor in USP18-mediated suppression of type I interferon signaling., 2017, 24(3): 279–289.
[23] Liu NX, Pang XX, Zhang H, Ji P. The cGAS-STING pathway in bacterial infection and bacterial immunity., 2021, 12: 814709.
[24] Bomfim CCB, Fisher L, Amaral EP, Mittereder L, McCann K, Correa AAS, Namasivayam S, Swamydas M, Moayeri M, Weiss JM, Chari R, McVicar DW, Costa DL, D'Império Lima MR, Sher A. Mycobacterium tuberculosis induces Irg1 in murine macrophages by a pathway involving both TLR-2 and STING/IFNAR signaling and requiring bacterial phagocytosis., 2022, 12: 862582.
[25] Huijser E, Bodewes ILA, Lourens MS, van Helden- Meeuwsen CG, van den Bosch TPP, Grashof DGB, van de Werken HJG, Lopes AP, van Roon JAG, van Daele PLA, Brkic Z, Dik WA, Versnel MA. Hyperresponsive cytosolic DNA-sensing pathway in monocytes from primary Sjögren's syndrome., 2022, 61(8): 3491–3496.
[26] Zhang M, Zhang MX, Zhang Q, Zhu GF, Yuan L, Zhang DE, Zhu QY, Yao J, Shu HB, Zhong B. USP18 recruits USP20 to promote innate antiviral response through deubiquitinating STING/MITA., 2016, 26(12): 1302–1319.
[27] Yang ZF, Xian HF, Hu JJ, Tian S, Qin YF, Wang RF, Cui J. USP18 negatively regulates NF-kappaB signaling by targeting TAK1 and NEMO for deubiquitination through distinct mechanisms., 2015, 5: 12738.
[28] Choi YB, Shembade N, Parvatiyar K, Balachandran S, Harhaj EW. TAX1BP1 restrains virus-induced apoptosis by facilitating itch-mediated degradation of the mitochondrial adaptor MAVS., 2017, 37(1): e00422–16.
[29] White J, Suklabaidya S, Vo MT, Choi YB, Harhaj EW. Multifaceted roles of TAX1BP1 in autophagy., 2023, 19(1): 44–53.
[30] Hou FJ, Sun LJ, Zheng H, Skaug B, Jiang QX, Chen ZJJ. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response., 2011, 146(3): 448–461.
[31] LIU BY, GAO CJ. Regulation of MAVS activation through post-translational modifications., 2018, 50: 75–81.
[32] Ling L, Goeddel DV. T6BP, a TRAF6-interacting protein involved in IL-1 signaling., 2000, 97(17): 9567–9572.
[33] Hou JX, Han LL, Zhao Z, Liu HQ, Zhang L, Ma CH, Yi F, Liu BY, Zheng Y, Gao CJ. USP18 positively regulates innate antiviral immunity by promoting K63-linked polyubiquitination of MAVS., 2021, 12(1): 2970.
[34] Zhu GF, Badonyi M, Franklin L, Seabra L, Rice GI, Anne-Boland-Auge, Deleuze JF, El-Chehadeh S, Anheim M, de Saint-Martin A, Pellegrini S, Marsh JA, Crow YJ, El-Daher MT. Type I interferonopathy due to a homozygous loss-of-inhibitory function mutation in STAT2., 2023, 43(4): 808–818.
[35] Löchte S, Waichman S, Beutel O, You CJ, Piehler J. Live cell micropatterning reveals the dynamics of signaling complexes at the plasma membrane., 2014, 207(3): 407–418.
[36] Tsft J, Bogunovic D. The goldilocks zone of type I IFNs: lessons from human genetics., 2018, 201(12): 3479–3485.
[37] Zhang XQ, Bogunovic D, Payelle-Brogard B, Francois- Newton V, Speer SD, Yuan C, Volpi S, Li Z, Sanal O, Mansouri D, Tezcan I, Rice GI, Chen CY, Mansouri N, Mahdaviani SA, Itan Y, Boisson B, Okada S, Zeng L, Wang X, Jiang H, Liu WQ, Han TT, Liu DL, Ma T, Wang B, Liu MG, Liu JY, Wang QK, Yalnizoglu D, Radoshevich L, Uzé G, Gros P, Rozenberg F, Zhang SY, Jouanguy E, Bustamante J, García-Sastre A, Abel L, Lebon P, Notarangelo LD, Crow YJ, Boisson-Dupuis S, Casanova JL, Pellegrini S. Human intracellular ISG15 prevents interferon-alpha/beta over-amplification and auto-inflammation., 2015, 517(7532): 89–93.
[38] Bogunovic D, Byun M, Durfee LA, Abhyankar A, Sanal O, Mansouri D, Salem S, Radovanovic I, Grant AV, Adimi P, Mansouri N, Okada S, Bryant VL, Kong XF, Kreins A, Velez MM, Boisson B, Khalilzadeh S, Ozcelik U, Darazam IA, Schoggins JW, Rice CM, Al-Muhsen S, Behr M, Vogt G, Puel A, Bustamante J, Gros P, Huibregtse JM, Abel L, Boisson-Dupuis S, Casanova JL. Mycobacterial disease and impaired IFN-gamma immunity in humans with inherited ISG15 deficiency., 2012, 337(6102): 1684–1688.
[39] Meuwissen ME, Schot R, Buta S, Oudesluijs G, Tinschert S, Speer SD, Li Z, van Unen L, Heijsman D, Goldmann T, Lequin MH, Kros JM, Stam W, Hermann M, Willemsen R, Brouwer RWW, Van IJcken WFJ, Martin-Fernandez M, de Coo I, Dudink J, de Vries FAT, Bertoli Avella A, Prinz M, Crow YJ, Verheijen FW, Pellegrini S, Bogunovic D, Mancini GMS. Human USP18 deficiency underlies type 1 interferonopathy leading to severe pseudo-TORCH syndrome., 2016, 213(7): 1163–1174.
[40] Alsohime F, Martin-Fernandez M, Temsah MH, Alabdulhafid M, Le Voyer T, Alghamdi M, Qiu X, Alotaibi N, Alkahtani A, Buta S, Jouanguy E, Al-Eyadhy A, Gruber C, Hasan GM, Bashiri FA, Halwani R, Hassan HH, Al-Muhsen S, Alkhamis N, Alsum Z, Casanova JL, Bustamante J, Bogunovic D, Alangari AA. JAK inhibitor therapy in a child with inherited USP18 deficiency., 2020, 382(3): 256–265.
[41] Muglia Amancio A, Mittereder L, Carletti A, Tosh KW, Green D, Antonelli LR, Gazzinelli RT, Sher A, Jankovic D. IFNs reset the differential capacity of human monocyte subsets to produce IL-12 in response to microbial stimulation., 2021, 206(7): 1642–1652.
[42] Liu XK, Li HX, Zhong B, Blonska M, Gorjestani S, Yan M, Tian Q, Zhang DE, Lin X, Dong C. USP18 inhibits NF-kappa B and NFAT activation during Th17 differentiation by deubiquitinating the TAK1-TAB1 complex., 2013, 210(8): 1575–1590.
[43] Martin-Fernandez M, Buta S, Le Voyer T, Li Z, Dynesen LT, Vuillier F, Franklin L, Ailal F, Muglia Amancio A, Malle L, Gruber C, Benhsaien I, Altman J, Taft J, Deswarte C, Roynard M, Nieto-Patlan A, Moriya K, Rosain J, Boddaert N, Bousfiha A, Crow YJ, Jankovic D, Sher A, Casanova JL, Pellegrini S, Bustamante J, Bogunovic D. A partial form of inherited human USP18 deficiency underlies infection and inflammation., 2022, 219(4): e20211273.
[44] Chen LM, Li SL, McGilvray I. The ISG15/USP18 ubiquitin-like pathway (ISGylation system) in hepatitis C virus infection and resistance to interferon therapy., 2011, 43(10): 1427–1431.
[45] Yan M, Luo JK, Ritchie KJ, Sakai I, Takeuchi K, Ren R, Zhang DE. Ubp43 regulates BCR-ABL leukemogenesis via the type 1 interferon receptor signaling., 2007, 110(1): 305–312.
[46] Pinto-Fernandez A, Salio M, Partridge T, Chen JZ, Vere G, Greenwood H, Olie CS, Damianou A, Scott HC, Pegg HJ, Chiarenza A, Díaz-Saez L, Smith P, Gonzalez-Lopez C, Patel B, Anderton E, Jones N, Hammonds TR, Huber K, Muschel R, Borrow P, Cerundolo V, Kessler BM. Deletion of the deISGylating enzyme USP18 enhances tumour cell antigenicity and radiosensitivity., 2021, 124(4): 817–830.
[47] Yim HY, Park C, Lee YD, Arimoto K, Jeon R, Baek SH, Zhang DE, Kim HH, Kim KI. Elevated response to type I IFN enhances RANKL-mediated osteoclastogenesis in usp18-knockout mice., 2016, 196(9): 3887–3895.
[48] Wang YQ, Mei YX, Song YS, Bachus C, Sun CX, Sheshbaradaran H, Glogauer M. AP-002: a novel inhibitor of osteoclast differentiation and function without disruption of osteogenesis., 2020, 889: 173613.
[49] Kar P, Millo T, Saha S, Mahtab S, Agarwal S, Goswami R. Osteogenic mechanisms of basal ganglia calcification and its ex vivo model in the hypoparathyroid milieu., 2021, 162(4): bqab024.
USP18-mediated protein deISGylation and its role in tuberculosis and other infectious diseases
Qi’ao Zhang1, Zilu Wang1, Peibo Li2, Jianping Xie1,2
The transcription of interferon-stimulated gene 15 () is induced by type I interferons. ISG15 can covalently modify target proteins through the sequential action of enzymesE1, E2, and E3, a process known as ISGylation. The ISGylation of host proteins is widely involved in immune responses, such as host antiviral defence. Ubiquitin-specific protease 18 (USP18), as a deubiquitinase (DUB), can remove ISG15 conjugated to target proteins and inhibit host immune responses by suppressing the type I interferon signaling. The dynamic balance between ISGylation and deISGylation mediated by ISG15 or USP18 respectively plays a significant role in the tuberculosis. Furthermore, similar to ISG15, USP18 is extensively involved in virus-host interaction. In this review, we summarize the roles of ISGylation and deISGylation in tuberculosis and other important diseases mediated by ISG15 and USP18 respectively, underlying regulator network. Further studies in this aspect will inspire new host-targeted strategies to control important diseases such as tuberculosis.
USP18; ISG15; tuberculosis; interferon
2023-07-07;
2023-10-17;
2023-11-01
国家自然科学基金项目(编号:82211530059, 82072246)资助[Supported by the National Natural Science Foundation of China (Nos. 82211530059, 82072246)]
张其奥,硕士研究生,专业方向:微生物感染与免疫。E-mail: zhqiao67@icloud.com
谢建平,博士,研究员,研究方向:结核分枝杆菌等重要病原致病耐药机理与新防控措施研发。E-mail: georgex@swu.edu.cn
10.16288/j.yczz.23-185
(责任编委: 张天宇)
猜你喜欢
中老年保健(2022年1期)2022-08-17
云南医药(2021年3期)2021-07-21
兽医导刊(2019年1期)2019-02-21
猪业科学(2018年8期)2018-09-28
长春中医药大学学报(2017年1期)2017-04-16
浙江大学学报(农业与生命科学版)(2015年4期)2015-12-15
中国医学科学院学报(2015年5期)2015-03-01
中国医药导报(2015年24期)2015-02-28
现代检验医学杂志(2015年2期)2015-02-06
中成药(2014年11期)2014-02-28
