
m6A修饰对于肿瘤在传统化疗和新型靶向治疗耐药中的作用研究进展*
2023-11-22 02:01郑思晴王昊
临床输血与检验 2023年5期
郑思晴 王昊
1安徽医科大学药学院,安徽合肥 230000;2中国科学技术大学附属第一医院(安徽省立医院)检验科,安徽合肥 230001
在哺乳动物信使RNA(mRNA)和非编码RNA(ncRNA)中发现的100多种RNA修饰类型中,腺苷N6位置的甲基化(N6-methyladenosine,m6A)被报道为是含量最丰富的类型[1-2]。m6A测序和CLIP测序等先进技术发现m6A甲基化针对的是共识序列DRACH (D=G,A或U;R=G或A;H=A,C或U),主要富集于mRNA的编码序列(CDS)和3'UTR区,以及其他种类的RNA上[1,3]。m6A甲基化已被证实在决定RNA命运中起着至关重要的作用,包括剪接、核输出、翻译、稳定和降解,并进一步调控基因表达和细胞表型[4-6]。m6A甲基化失调参与了许多病理过程,包括肿瘤的发生、进展、转移和治疗耐药[7-8]。
在全球范围内,癌症仍然是公共卫生的头号威胁,并对社会造成严重的经济负担[9]。由于存在显著的异质性,癌细胞经常表现出治疗耐药性,这是癌症治疗的主要障碍[10]。化学耐药获得机制包括细胞积累减少、解毒增加、DNA修复增加、细胞凋亡减少和自噬等[11]。不断有研究表明m6A修饰可通过这些方式调节肿瘤耐药[12-14]。
综上所述,阐明m6A甲基化修饰对治疗耐药的影响,对于探索克服耐药的潜在策略,开发更有效的治疗靶点,最终优化癌症治疗具有深远的意义[4,15]。在本文中,我们系统地总结了最新的研究成果,为m6A甲基化修饰调控的耐药形成机制提供深入的见解,旨在促进潜在治疗策略的开发。
1 m6A的甲基化和去甲基化
m6A甲基化主要由甲基转移酶复合物(methyltransferase complex,MTC)催化,MTC是由甲基转移酶样3(methyltransferase-like3,METTL3)和METTL14组成的异源二聚体,其中METTL3发挥催化作用,METTL14提供结构支持并识别靶RNA[16]。Wilms肿瘤1相关蛋白(Wilms tumor 1-associated protein,WTAP)、vir样m6A甲基转移酶相关蛋白(vir-like m6A methyltransferase associated,VIRMA/KIAA1429)、RNA结合基元蛋白15(RNA-binding motif protein15,RBM15)和锌指CCCH结构域蛋白13(zinc finger CCCH domaincontaining protein13,ZC3H13)作为辅助因子,将MTC定位到靶位点并启动甲基化[17]。
m6A去甲基化由肥胖相关蛋白(obesityassociated protein,FTO)和ALKB同源物5 (ALKB homology5,ALKBH5)两种去甲基化酶执行[18-19]。FTO是第一个被发现的m6A去甲基化酶,主要影响mRNA的稳定性、翻译和剪接[20-21]。ALKBH5作为FTO的同源物,可以调节RNA剪接、输出和降解[22]。
m6A结合蛋白能够特异性识别并结合m6A位点,控制RNA命运,实现基因表达和生物学表型的表观遗传调控。YT521-B同源(YT521-B homology,YTH)蛋白,包括YTHDC1-2和YTHDF1-3,广泛参与RNA代谢。已证实YTHDC1促进RNA剪接、核输出和衰变[5,23-24],YTHDC2有助于提高翻译效率和mRNA衰变[25-26]。YTHDF1通过与真核起始因子3(eukaryotic initiation factor3,eIF3)相互作用促进翻译起始,YTHDF3不仅与YTHDF1协同促进翻译,还与YTHDF2协同诱导mRNA降解[27-28]。胰岛素样生长因子2 mRNA结合蛋白家族(insulin-like growth factor 2 mRNA binding protein Family,IGF2BP1-3)通过K同源结构域识别m6A靶点,促进RNA的稳定性和翻译[29]。异质核核糖核蛋白(heterogeneous nuclear ribonucleoproteins,HNRNPs)被证实可引发选择性剪接并介导共翻译调节[30]。此外,脆性X型智力迟钝蛋白(fragile X mental retarded proteins,FMRPs)在RNA输出中是不可或缺的[31]。
甲基转移酶、去甲基转移酶和m6A结合蛋白共同催化、去除和识别m6A甲基化,建立可逆的动态平衡。除mRNA外,包括tRNA、rRNA、miRNA、lncRNA和circRNA在内的ncRNA都可以被m6A修饰,从而影响RNA的剪接、翻译、成熟、转运、定位以及RNA与蛋白的相互作用。
2 m6A和化疗耐药
化疗是临床治疗癌症最有效的策略之一,特别是对于不能进行手术治疗的晚期恶性肿瘤患者[32]。然而对化疗药物的耐药极大地限制了整体治疗效率,并逐渐成为日益严峻的临床问题[11]。值得注意的是,越来越多的证据强调m6A修饰是肿瘤对化疗反应的潜在决定因素(图1,图2)。

图1 METTL3通过靶向不同分子对传统化疗药物耐药的促进或抑制作用

图2 METTL3以外的其他m6A调控分子(WTAP、VIRMA、YTHDF1、YTHDF2、YTHDF3、IGF2BP3、METTL14、ALKBH5、FTO)通过靶向不同分子对传统化疗药物耐药的促进或抑制作用
2.1 铂类药物
第一代铂类药物顺铂于1965年被FDA批准用于睾丸癌,随后第二代卡铂和第三代奥沙利铂在世界范围内被用于多种类型的肿瘤[33]。然而,铂类药物的耐药通常在短时间内出现,m6A已被证实可调节顺铂(cisplatin,DDP)和奥沙利铂(oxaliplatin,OXA)的耐药。
在非小细胞肺癌(non-small cell lung cancer,NSCLC)中,METTL3是否促进或抑制顺铂耐药尚无一致结论。研究发现,METTL3通过提高AKT丝氨酸/苏氨酸激酶1 (AKT serine/threonine kinase1,AKT1)蛋白的表达而导致顺铂不敏感,且METTL3在耐药NSCLC样本中表达上调并与生存率低呈正相关[34]。然而,LING等人发现异丙酚通过促进METTL3介导的m6A甲基化增强肿瘤对顺铂的敏感性。异丙酚增加了pri-miR-486-5p上m6A的富集,促进其成熟,并进一步使Ras associated protein-1(RAP1)-NF-kappa B(NF-κB)轴失活,而METTL3的敲低则逆转了这一促进作用[35]。此外,METTL3受富集于顺铂耐药NSCLC的外泌体中miR-4443的负调控。减少的METTL3进一步上调铁下垂抑制蛋白1 (ferroptosis suppressor protein1,FSP1),减少DDP诱导的铁死亡,从而产生化学耐药[36]。
在胃癌(gastric cancer,GC)中,既往研究一致证实METTL3促进了肿瘤对铂类药物的耐受。据报道,METTL3将Rho GTPase激活蛋白5 (Rho GTPase activating protein 5,ARHGAP5) mRNA甲基化促进其翻译,从而使肿瘤对顺铂、盐酸阿霉素和5-氟尿嘧啶(5-Fu)等化疗药物产生耐药性[37]。此外,在CD133+胃癌干细胞中,METTL3在奥沙利铂耐药中起决定性作用。在机制上,上调的METTL3可以通过募集YTHDF1到3'UTR,增强PARP1mRNA的稳定性,PARP1有助于修复药物诱导的DNA损伤[12]。YTHDF2参与胱硫氨酸β合成酶mRNA不稳定lncRNA(cystathionineβ-synthase mRNA-destabilizing lncRNA,CBSLR)诱导的CBS mRNA的失稳,下调CBS可降低酰基辅酶A合成酶长链家族4(Acyl-CoA Synthetase Long Chain Family Member 4,ACSL4)的甲基化,在体内和体外保护胃癌细胞免于铁凋亡,从而导致化疗反应较差[38]。此外,武藏RNA结合蛋白2(musashi RNA binding protein2,MSI2)被认为是潜在的m6A“阅读者”,可以促进DDP耐药性。在机制上,LNC942通过抑制MSI2泛素化促进其表达。MSI2以依赖m6A的方式稳定c-Myc mRNA[39]。而LNC942先前报道通过招募METTL14稳定下游靶点,这表明m6A参与其中[40]。
在结直肠癌(colorectal cancer,CRC)中,METTL3被认为显著上调并维持奥沙利铂耐药。机制研究表明,METTL3可依赖IGF2BP1增加CBX8mRNA的稳定性,并促进富含亮氨酸重复序列G蛋白偶联受体5(leucine rich repeat containing G proteincoupled receptor5,LGR5)的转录,维持肿瘤的干性和化疗耐药[41]。METTL3触发m6A修饰肿瘤坏死因子受体相关因子(tumor necrosis factor receptorassociated factors,TRAF) mRNA,促进其降解,从而抑制坏死下垂,降低OXA敏感性。同时,M2肿瘤相关巨噬细胞(M2 tumor associated macrophages,TAMs)通过增强m6A甲基化促进OXA耐药形成[42]。在DDP耐药的CRC细胞中,过表达的YTHDF1通过结合谷氨酰胺酶1(glutaminase1,GLS1) mRNA的3'UTR促进 GLS1的蛋白合成。体外和体内实验中,抑制YTHDF1介导的谷氨酰胺代谢均可使结直肠癌细胞对顺铂敏感[43]。
在卵巢癌(ovarian cancer OC)中,ALKBH5在化疗耐药获得过程中发挥重要作用。ALKBH5通过与上游转录因子同源盒A10(upstream transcription factor homeobox A10,HOXA10)形成环,随后通过m6A去甲基化共同激活Janus 激酶2(Janus kinase 2,JAK2)/信号转换器和转录激活因子3(activator of transcription3,STAT3)信号通路,从而促进化疗耐药[44]。三结构域蛋白(tripartite motif-containing protein,TRIM29)被认为是卵巢癌中与恶性特征相关的致癌基因,YTHDF1结合TRIM29 mRNA上的m6A位点,促进其翻译,增强DDP抗性[45]。
为了开发更好和更有效的癌症治疗方法,研究人员已经开始阐明顺铂耐药的潜在机制。表观遗传修饰已被证明与化疗耐药有关,而表观遗传生物标志物,如尿肿瘤DNA甲基化测定(urine tumor DNA methylation assay,utMeMA),已被用于膀胱癌患者筛查或监测[46]。
2.2 替莫唑胺
替莫唑胺(temozolomide,TMZ)是铂类药物之外的另一种烷化剂。也是治疗晚期胶质瘤主要的一线化疗药物,联合放疗作为标准治疗方案[47]。然而,该方案在胶质母细胞瘤(glioblastoma,GBM)中的总体临床疗效仍然令人沮丧,因为TMZ治疗存在固有或诱导的耐药性[48]。结果表明,在体外和体内,METTL3水平在GBM中升高,并促进肿瘤对TMZ的耐药。在机制上,METTL3介导的m6A修饰可增强O6-甲基鸟嘌呤-DNA甲基转移酶(O6-methylguanine-DNA methyltransferase,MGMT)和烷基嘌呤-DNA-N-糖基化酶(alkylpurine-DNAN-glycosylase,APNG)的表达,从而修复DNA损伤[48]。据报道,去甲基化酶FTO也增强了GBM对TMZ的抗性。FTO介导的去甲基化保护磷酸肌苷依赖性激酶-1(phosphoinositide dependent kinase-1,PDK1) mRNA免于降解,而X无活性特异性转录物(X-inactive specific transcript,JPX)显著调节FTO/PDK1相互作用,导致GBM的进展和化疗耐药[49]。值得注意的是,FTO抑制剂R-2HG与TMZ具有协同作用[50]。此外,circ-0072083可以通过负调控miR-1252-5p提高ALKBH5表达水平,从而减少m6A修饰依赖性降解促进NANOG 同源盒(Nanog Homeobox,NANOG)表达,而NANOG是化疗耐药的重要生物标志物[51]。
2.3 吉西他滨
吉西他滨(gemcitabine,GEM)是一种嘧啶类似物,临床上广泛用于多种肿瘤的治疗。它的应用彻底改变了胰腺癌(pancreatic cancer,PC)的治疗,使患者生存率提高到20%[52]。然而,GEM的化学耐药已成为一个紧迫的问题,且受到m6A修饰的调控。METTL3在调节GEM敏感性中起积极作用。m6A甲基化维持胰腺癌细胞中DBH-反义RNA(DBH Antisense RNA,DBH-AS1)的RNA稳定性,通过分离miR-3163使胰腺癌细胞对GEM敏感。特别是在对GEM耐药的胰腺癌组织和细胞中,DBH-AS1表达下调,且与METTL3的低表达密切相关[53]。与METTL3相反,METTL14在GEM耐药的胰腺癌细胞中被转录因子p65诱导上调,并在下游增强胞苷脱氨酶(cytidine deaminase,CDA)的表达,使GEM失活。抑制METTL14在体内和体外均能明显增强GEM的敏感性,表明METTL14是改善胰腺癌化疗效果的有希望的靶点[54]。
2.4 氟尿嘧啶
氟尿嘧啶(fluorouracil,5-FU)是尿嘧啶的类似物,可与核酸结合,干扰核苷酸代谢[55]。它已被广泛应用于常见的化疗方案,包括双PF(顺铂和5-FU),用于多种肿瘤的治疗[56]。5-FU是结直肠癌姑息治疗和辅助治疗的首选,尽管其有效率不理想且耐药频繁[57]。一些研究已经确定METTL3是5-FU抵抗的驱动力。研究发现,m6A修饰可增强DiGeorge综合征关键区域8 (DiGeorge Syndrome Critical Region8,DGCR8)对pri-miR181d的识别,从而促进miR181d5p过程。升高的miR181d5p与神经钙素δ(neurocalcin delta,NCALD) mRNA的3'UTR相互作用抑制NCALD的表达,诱导结直肠癌细胞对5-FU的耐药性[58]。同样,METTL3可以增强依赖于IGF2BP1的LBX2反义RNA1(LBX2 Antisense RNA 1,LBX2-AS1)的稳定性,上调的LBX2-AS1通过海绵化miR-422a进一步提高AKT1水平,与结直肠癌患者对5-FU的不良反应相关[59]。
LIN等人研究表明临床CRC组织中SIVA1细胞凋亡诱导因子(SIVA1 Apoptosis Inducing Factor,SIVA1)1水平与FTO水平呈负相关。值得注意的是,FTO的抑制通过FTO-SIVA1轴显著降低了5-FU耐药的CRC细胞对5-FU的耐受性,SIVA1的缺失可以恢复CRC细胞中依赖于m6A的5-FU敏感性。总之,FTO作为一种m6A去甲基化酶在CRC细胞化疗耐药中发挥着关键作用,并提示抑制FTO可能恢复化疗耐药CRC细胞对5-FU的敏感性[60]。NISHIZAWA等人提出,抑制c-Myc驱动的YTHDF1转激活可抑制结直肠癌的进展,并引发对OXA和5-FU等抗癌药物的敏感化。高水平的YTHDF1患者的总生存期明显较差[61]。另一项研究表明,miR-136-5p下调YTHDF1,抑制肿瘤进展和对5-FU和OXA的化疗耐药,而miR-136-5p被circPTK2靶向,在结直肠癌细胞系和组织中下降[62]。
2.5 阿霉素/多柔比星
多柔比星(doxorubicin,DOX)是一种蒽环类抗生素,通过嵌入DNA并随后破坏DNA修复[63]或通过氧化应激[64]发挥抗肿瘤作用。DOX仍然是早期和晚期乳腺癌(breast cancer,BC)以及许多其他恶性肿瘤的主要治疗药物之一。除了致命的心脏毒性外,耐药性是影响化疗效果的主要障碍。
一些研究已经阐明了METTL3在乳腺癌中引发DOX耐药的不同途径。LI等人观察到METTL3在DOX耐药乳腺癌细胞中高表达。他们认为METTL3促进转移相关肺腺癌转录物1(metastasis associated lung adenocarcinoma transcript1,MALAT1)的表达,MALAT1募集E2F转录因子1(E2F transcription factor1,E2F1)激活前梯度2 (anterior gradient2,AGR2)的转录,从而使乳腺癌细胞对DOX脱敏[65]。METTL3可促进pri-miR-221-3p成熟,miR-221-3p进一步抑制同源结构域相互作用蛋白激酶2(Homeodomain Interacting Protein Kinase 2,HIPK2)并上调下游的Che-1,导致多药耐药蛋白(multidrug resistance protein,MDRP)和乳腺癌耐药蛋白(breast cancer resistance protein,BCRP)升高,表明化疗耐药增强[66]。METTL3介导的m6A修饰通过触发雌激素受体相关受体γ (estrogen receptor related receptor γ,ERRγ)pre-mRNA剪接促进ERRγ的表达,ERRγ与p65相互作用促进ATP结合盒亚家族B成员1(ATP Binding Cassette Subfamily B Member 1,ABCB1)转录,编码P-gp,降低对紫杉醇(Taxol,Tax)和DOX的敏感性。ERRγ/p65复合物还可结合肉碱棕榈酰转移酶1B(Carnitine Palmitoyl transferase 1B,CPT1B)启动子,增加其表达,加速脂肪酸β-氧化(fatty acidβ-oxidation,FAO),从而促进耐药[67]。此外,脂肪酸的重新合成被认为与乳腺癌的化疗耐药有关。抑制FTO显著减弱脂肪酸合成,这明显使乳腺癌细胞对化疗敏感[68]。
其他m6A调节剂对于乳腺癌对DOX的耐药也有影响。WTAP增强lncRNA DLGAP1反义RNA 1(DLGAP1 antisense RNA1,DLGAP1-AS1)的稳定性,DLGAP1-AS1过表达促进体外化疗耐药,与乳腺癌患者预后不良相关。此外,DLGAP1-AS1通过海绵化miR-299-3p来缓解其对WTAP的抑制,从而激活WTAP,形成一个反馈回路[69]。WANG等人研究发现,FTO和STAT3在DOX耐药的乳腺癌细胞中高表达,STAT3直接结合FTO的启动子区域调节其转录。FTO敲低可增加对DOX的化学敏感性,逆转STAT3过表达的相反作用[70]。
除乳腺癌外,m6A甲基化也被发现能够调节其他癌症的DOX耐药。YANG等研究发现,结直肠癌中IGF2BP3上调可通过增加mRNA稳定性促进ABCB1表达,从而诱导体外和体内对DOX的耐药[71]。在骨肉瘤(osteosarcoma,OS)中,当METTL3和METTL14被消融时,m6A修饰在三元基序家族蛋白7(Tripartite Motif Containing Protein7,TRIM7)mRNA上的减少将导致YTHDF2无法结合并降解该mRNA,从而使TRIM7的表达水平上调。TRIM7作为一种泛素连接酶,通过泛素化来抑制BRMS1,使其在体外和体内对阿霉素(Adriamycin,ADR)和甲氨蝶呤(methotrexate,MTX)的耐药性增强[72]。在DOX耐药的慢性髓细胞性白血病(chronic myelocytic leukemia,CML)细胞中,LINC00470通过METTL3修饰的甲基化促进磷酸酶和紧张素同源物(phosphatase and tension homology,PTEN)mRNA的降解,从而通过抑制Notch和PI3K-AKTMtor信号通路发挥肿瘤抑制作用。敲低METTL3可挽救PTEN表达,从而抑制白血病细胞活力,恢复化疗敏感性[73]。
2.6 其他化疗药物
WANG等人已经证明了脂肪细胞来源的外泌体在保护多发性骨髓瘤(multiple myeloma,MM)细胞免受化疗药物(如硼替佐米、美法兰和卡氟佐米)诱导的细胞凋亡方面的作用。METTL7A促进IncRNAs富集到外泌体中发挥致癌作用,MM细胞通过EZH-2介导的METTL7A甲基化增强了这一过程。这一发现提示了通过阻断这种外泌体介导的恶性循环来改善耐药性的潜在策略[74]。在急性髓性白血病(acute myeloid leukemia,AML)中,METTL3在耐药AML细胞中表达上调。METTL3可以通过促进MYC的表达来促进对阿糖胞苷(cytarabine,Ara-C)的化疗耐药[75]。然而,AML骨髓间充质干细胞(bone marrow mesenchymal stem cells,MSCs)的整体m6A水平和METTL3的表达降低,并且METTL3的过表达使MSCs对青霉素和链霉素敏感。下调的METTL3上调AKT水平,提高脂肪生成,从而促进化疗耐药[76]。
在AML患者中,移植后复发非常常见,是治疗失败和死亡的主要原因。最近,糖蛋白-A为主的重复序列 (glycoprotein-A repetitions predominant,GARP)被发现是一种潜在的转化生长因子-β(transforming growth factor-β1,TGF-β)结合蛋白,在调节TGF-β1的生物利用度中起着关键作用[77]。WANG等人研究发现GARP介导的TGF-β1活性释放在异源造血干细胞移植后早期复发的AML患者中诱导骨髓自然杀伤 (bone marrow Natural killer,BMNK)细胞功能障碍,而galunisertib抑制TGF-β1信号通路可以在体外和体内恢复NK细胞的抗肿瘤活性。因此,TGF-β1信号抑制剂可能通过恢复NK细胞对肿瘤具有治疗作用。然而,需要进一步的研究来确定galunisertib治疗AML患者的剂量、安全性和有效性[77]。
LIU等人构建了一个基于YTHDF3的新型模型,用于有效预测几种乳腺癌治疗剂的敏感性,包括阿霉素、紫杉醇、甲氨蝶呤和长春瑞滨[78]。ZHANG等基于7个m6A调节因子构建了预测小细胞肺癌化疗获益的有效分类器。体外实验表明,ZCCHC4、G3BP1和RBMX可能是克服小细胞肺癌化疗耐药的潜在治疗靶点[79]。
3 m6A和靶向治疗耐药
靶向治疗是一种突破性的治疗方法,通过干扰特定分子来阻止癌症的生长、进展和转移。一系列靶向药物已被FDA批准,在治疗包括肝癌、结直肠癌、肺癌、乳腺癌和卵巢癌在内的各种癌症类型方面显示出良好的临床疗效。然而,对靶向治疗的耐药经常出现,主要是由于靶突变、旁路改变和细胞可塑性[80]。同时,m6A甲基化已被证实可影响靶向分子或途径,调节治疗耐药性(图3,图4)。

图3 METTL3通过靶向不同分子对新型靶向药物耐药的促进或抑制作用
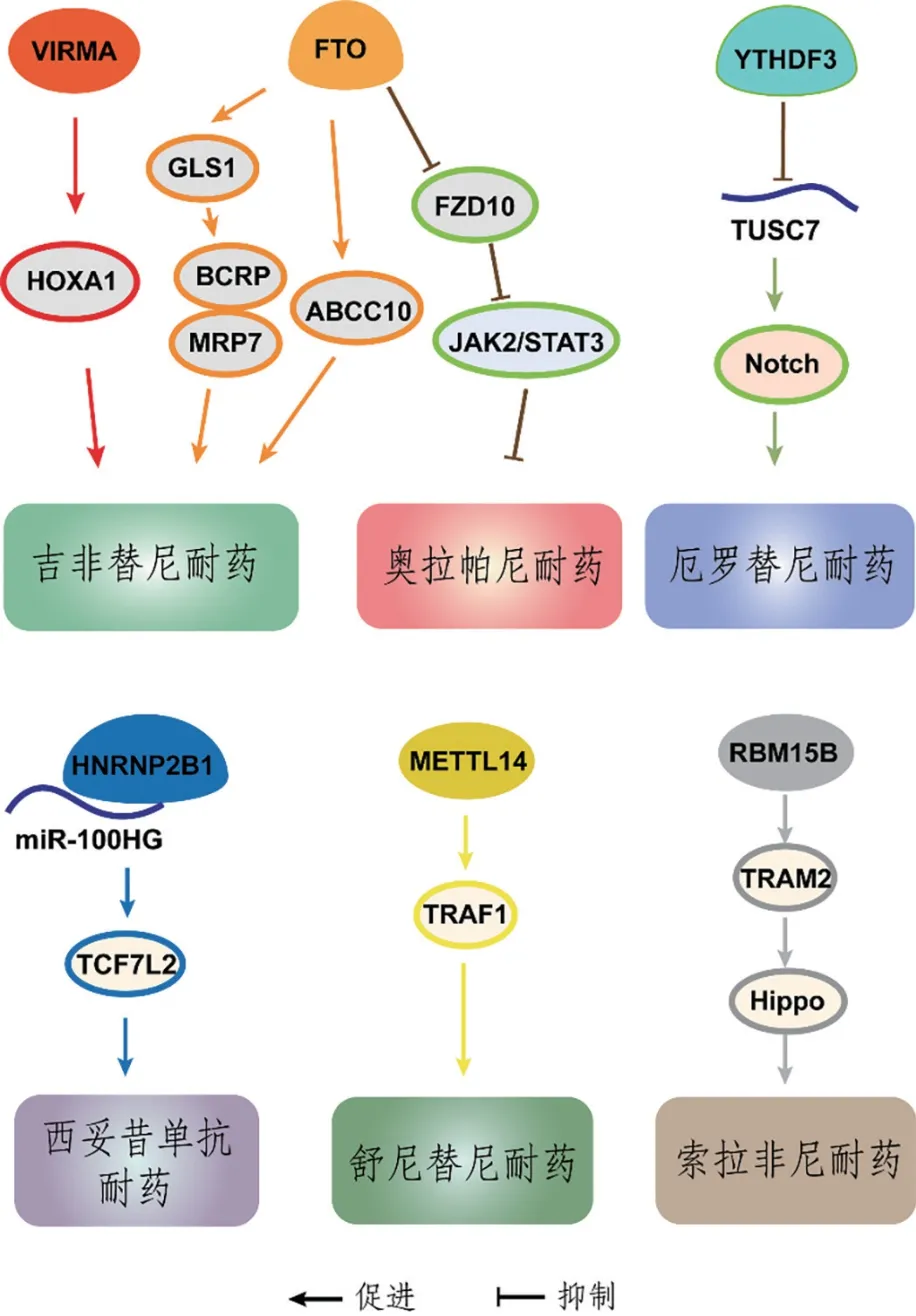
图4 METTL3以外的其他m6A调控分子(VIRMA、FTO、YTHDF3、HNRNP2B1、METTL14、RBM15B)通过靶向不同分子对新型靶向药物耐药的促进或抑制作用
3.1 索拉非尼
索拉非尼是一种多靶点酪氨酸激酶抑制剂(tyrosine kinase inhibitor,TKI),可通过抑制Ras/Raf/MEK/ERK信号通路抑制癌细胞增殖,并可靶向血管内皮生长因子受体2(vascular endothelial growth factor receptor,VEGFR2)、血小板衍生生长因子受体(platelet-derived growth factor receptor,PDGFR-β)等受体抑制血管生成[81]。索拉非尼的应用显著改善了晚期肝细胞癌(hepatocellular carcinoma,HCC)患者的预后[82]。
对于METTL3和METTL14,关于它们在索拉非尼耐药中的调节作用有相互矛盾的发现。在促进作用方面,高表达的METTL3显著增强了长链非编码RNA 双同源盒A假基因8(IncRNA Double homeobox A pseudogene 8,IncRNADUXAP8)的稳定性,后者通过ceRNA机制与miR-584-5p竞争性结合,从而促进MAPK1表达,激活MAPK/ERK通路,促进肝细胞癌耐药[83]。METTL3可上调NIFK-AS1,NIFK-AS1抑制药物转运体有机阴离子转运多肽1B1(organic anion-transporting poly peptide 1B1,OATP1B1)和OATP1B3的表达,抑制索拉非尼摄取,从而使肝细胞癌细胞脱敏。低水平NIFK-AS1的肝细胞癌患者表现出治疗的益处[84]。此外,m6A-circRNA相互作用在维持耐药性中被发现。METTL3/14通过增加其稳定性来促进circRNA SORE的表达,其作为海绵隔离miR-103a2-5p和miR-660-3p以激活Wnt/β-catenin通路,从而促进耐药[85]。相反,LIN等人提出敲低METLL3可通过FOXO3介导的自噬促进耐药。METTL3缺失使FOXO3mRNA 3'UTR处的m6A水平减弱,抑制YTHDF1介导的稳定,进而下调FOXO3诱导一系列自噬相关基因的转录,增强肝癌细胞系的自噬[86]。此外,机制研究表明,METTL14可以通过IGF2BPs增强HNF3γ mRNA的稳定性,从而诱导OATP1B1和OATP1B3表达的反激活,肝癌细胞中METTL14的表达减少有助于产生耐药[87]。除了两种主要的甲基转移酶外,另一种甲基转移酶RBM15B也被认为能促进肝癌患者对索拉非尼的耐药性。RBM15B与TRAM2mRNA相互作用可增强其稳定性,增加的TRAM2促进YAP和TAZ的表达和激活,激活Hippo信号通路。RBM15B和TRAM2的下调可显著逆转耐药,而过表达则产生相反的效果[88]。
3.2 吉非替尼
表皮生长因子受体(epidermal growth factor receptor,EGFR)过表达在实体瘤中普遍存在,并与肿瘤的发生、进展和侵袭有关。EGFR-TKI治疗为EGFR激活突变的NSCLC患者提供了巨大的治疗效果[89]。对于EGFR-外显子19框内缺失或外显子21单点突变的肺癌患者,标准的一线治疗是第一代(吉非替尼、厄洛替尼)或第二代(阿法替尼)TKIs[90]。
在NSCLC中,已有证据表明METTL3在获得吉非替尼耐药中发挥了积极作用。METTL3通过稳定SNHG17mRNA诱导其上调,SNHG17将EZH2募集到大肿瘤抑制激酶2 (large tumor suppressor kinase2,LATS2)的启动子区域抑制其表达,通过表观遗传抑制LATS2促进肺腺癌患者对吉非替尼的耐药[91]。此外,METTL3可以通过增加自噬来促进耐药。过表达的METTL3上调了自噬通路的ATG5、ATG7等基因,而β-榄香烯高选择性地抑制了METTL3的表达,从而通过破坏自噬逆转耐药性[92]。此外,TANG等人报道了甲基转移酶KIAA1429在耐药NSCLC细胞中的表达增加,并伴有m6A的高富集。KIAA1429在体外通过靶向HOXA1mRNA的3'UTR增强其稳定性,以依赖于m6A的方式促进耐药性[93]。LIN等人阐明了病毒样m6A甲基转移酶相关蛋白KIAA1429基因的下调以m6A甲基化依赖的方式抑制MAP3K2的表达,抑制肺腺癌(lung adenocarcinoma,LUAD)细胞的发展,抑制吉非替尼耐药HCC827细胞的生长。KIAA1429正向调控MAP3K2的表达,激活JNK/ MAPK通路,促进耐吉非替尼HCC827细胞的耐药。因此m6AKIAA1429介导的LUAD细胞吉非替尼耐药机制是通过激活JNK/ MAPK信号通路实现的。这些发现为吉非替尼耐药的LUAD患者提供了潜在的分子治疗和临床治疗靶点[94]。
FTO也被发现在NSCLC中表达上调,有利于对吉非替尼的耐药。吉非替尼耐药NSCLC患者血清外泌体中FTO表达升高,m6A水平降低,FTO的缺失显著增强了敏感性。FTO通过抑制YTHDF2介导的mRNA衰变促进ABCC10的表达,促进体外和体内的耐药性[95]。值得注意的是,FTO抑制剂甲氯芬酸(meclofenamic acid,MA)通过抑制FTO介导的m6A修饰MYC的去甲基化,并进一步下调膜药物流蛋白、BCRP和MRP-7来克服耐药性[96]。
3.3 其他靶向治疗药物
厄洛替尼是第一代EGFR抑制剂,与吉非替尼具有相似的分子结构和药理机制。在肺癌中,YTHDF2介导的lncRNATUSC7抑制会引发对厄洛替尼的耐药,因为TUSC7可以海绵化miR-146a并抑制Notch信号从而降低癌症进展并提高对厄洛替尼的敏感性[97]。奥西替尼是第一个被批准用于转移性EGFR-突变型NSCLC的第三代EGFR-TKIs。在奥昔替尼耐药的肺癌细胞中,二甲双胍可以通过促进DNA甲基转移酶3a/b与METTL3启动子的结合而上调METTL3。METTL3介导的m6A通过NKAP和HNRNPA2B1显著促进了pre-Let-7b的成熟,从而使Notch信号失活并重新吸引奥西替尼治疗[98]。
阿帕替尼是一种靶向VEGFR-2抗血管生成的选择性TKI,应用于胃癌、肝癌、NSCLC等[99]。在肝癌中,s-腺苷同型半胱氨酸或siRNA诱导的METTL3抑制可激活p53,通过凋亡进一步使肝癌细胞对阿帕替尼敏感[100]。舒尼替尼是一种多靶点、抗血管生成的TKI,极大地提高了转移性肾细胞癌(renal cell carcinoma,RCC)患者的总生存率。由于METTL14介导的m6A修饰以依赖IGF2BP2的方式增强了TRAF1mRNA的稳定性,肾癌对舒尼替尼的耐药被认为与TRAF1的高表达水平有关[101]。
克唑替尼是一种靶向c-MET/ALK/ROS1的激酶抑制剂,用于ALK突变的NSCLC的一线治疗。在ALK无突变和c-MET高表达的NSCLC细胞系中,西达本胺可以通过降低METTL3和WTAP的表达来增强对克唑替尼的敏感性,降低c-MET mRNA上的m6A水平下调其表达,随后恢复对克唑替尼的反应,而且c-MET配体肝细胞生长因子(hepatocyte growth factor,HGF)可与西达本胺发挥协同作用[102]。聚(ADP -核糖)聚合酶抑制剂(Poly (ADP-ribose)polymerase inhibitors,PARPi)如奥拉帕尼,已被证明对BRCA1/2突变的上皮性卵巢癌(Epithelial ovarian cancer,EOC)患者有显著的治疗效果[103]。在BRCA突变的卵巢癌细胞中,FTO和ALKBH5的下调使FZD10转录本m6A水平升高,并通过IGF2BP2稳定FZD10转录本m6A水平,进一步刺激Wnt/β-catenin通路,导致对奥拉帕尼和卢卡帕尼产生耐药性[104]。
4 靶向m6A调节剂克服药物耐受性
随着对m6A修饰调控癌症治疗耐药的机制研究越来越多,靶向m6A来优化癌症治疗变得越来越有吸引力。研究发现m6A调节因子的特异性抑制剂已显示出相当有效的抗肿瘤作用,包括METTL3、FTO、ALKBH5、IGF2BP1的抑制剂[105],而METTL3激活剂的治疗作用尚未得到证实[106]。最近,ELIZA等人通过对25万种药物样化合物的高通量筛选,发现了一种高效、选择性的METTL3和METTL14的一级催化抑制剂,命名为STM2457。该抑制剂在临床前AML模型中显示出显著的抗白血病作用,证明靶向m6A基因是一种很有前景的癌症治疗策略[107]。WENG等人基于结构的虚拟筛选鉴定出IGF2BP2抑制剂NSC69557,通过高效液相色谱法纯化了该化合物并将对AML细胞活力/生长的抑制作用最强的小分子抑制剂命名为CWI1-2。CWI1-2在体外和体内均显示出良好的抗白血病作用[108]。此外,研究强调了几种特定抑制剂克服治疗耐药性的潜力,支持与现有治疗方法的联合应用。大黄酸(rhein)是第一个与FTO催化结构域竞争性结合的天然FTO抑制剂,并在白血病小鼠中显示出治疗效果[109]。非甾体抗炎药甲氯芬酸2(MA2)被发现可以抑制FTO,从而抑制胶质母细胞瘤的进展[110]。R-2-羟基戊二酸酯(R-2-hydroxyglutarate,R-2HG)通过抑制FTO在体外和体内抑制AML的进展,并与全反式维甲酸(all-trans retinoic acid,ATRA)、阿扎胞苷、地西他滨和柔红霉素等一系列一线化疗药物协同作用[50]。此外,R-HG在GBM细胞系中表现出生长抑制作用,并与TMZ协同作用[50]。随后,HUANG等人报道了三环苯甲酸FB23-2可抑制AML细胞的增殖并增强其分化/凋亡[111]。最近的研究表明,FB23类似物FB23-13a在体外和体内对AML细胞具有更强的抗增殖作用[112]。近年来的研究在AML和胶质瘤以外的更多肿瘤类型中取得了良好的进展。18097是一种小分子化合物,能显著抑制乳腺癌细胞的生长和定植[113]。值得注意的是,氧烷类FTO抑制剂FTO-43在胃癌、胶质母细胞瘤和AML模型中显示出与5-FU相当的抗肿瘤效力[114]。
肿瘤微环境(tumor microenvironment,TME)在癌症进展中起重要作用,并显著影响免疫治疗的效果[115]。最近的研究表明,肿瘤细胞中的m6A调节因子与免疫细胞反应和免疫检查点阻断(immune checkpoint blockade,ICBs)的有效性有关。肿瘤细胞中m6A修饰的改变会影响TME中炎性免疫细胞的浸润、激活和效应功能[116],因此靶向肿瘤细胞中的m6A,尤其是m6A调节因子,是改善癌症免疫治疗的一种有前景的策略。自然杀伤细胞(natural killer,NK)是一种先天淋巴免疫细胞,具有直接识别和杀伤癌细胞的能力,在肿瘤免疫监视中起着重要作用[117]。MAS等人首次报道了YTHDF2介导的m6A甲基化在NK细胞免疫中的多方面作用[118]。YTHDF2对于维持NK细胞稳态、成熟、白细胞介素(IL)-15介导的存活以及抗肿瘤和抗病毒活性至关重要,因为它调节了几个下游靶基因,包括转录信号换能器和激活因子5 (STAT5)、Eomes和Tardbp[118]。
SUN等人的研究发现m6A甲基转移酶METTL3通过靶向脱帽蛋白2(Decapping Protein 2,DCP2)调控Pink1-Parkin通路介导的小细胞肺癌(small cell lung cancer,SCLC)细胞线粒体自噬和线粒体损伤,从而促进SCLC患者化疗耐药[119]。最初的小分子METTL3抑制剂是通过高通量对接到SAM结合位点发现的,它确实是腺苷类似物[120]。然而,由于其结合混杂性和渗透性差,非核苷类抑制剂被开发出来。UZH1a是一种小分子METTL3抑制剂,对AML具有有效的生长抑制作用[121]。UZH1a的类似物UZH2在AML和前列腺癌细胞系中表现出抗肿瘤作用[122]。
总的来说,靶向m6A调节因子的药物作为一种有前景的治疗策略受到了广泛的关注。对于已证实能提高治疗效果的特异性抑制剂,还需进一步加强临床前研究。对于这些新药物,应探索其协同现有治疗方法和克服耐药的潜力。此外,还应努力开发更有效、更有选择性的抑制剂或激活剂。
5 m6A研究前景
近年来,m6A修饰在控制肿瘤治疗耐药中的调节作用越来越受到关注。靶向m6A修饰系统非常有利于提高癌症治疗效果,并在预防耐药和开发新的治疗策略方面具有重要的临床意义。
然而,随着这一领域的突破,相关的理论基础仍然存在矛盾和不确定性:(1)在同一种肿瘤中,m6A甲基转移酶和去甲基转移酶的表达方向相同,导致m6A浓度变化存在差异。例如,METTL3和FTO在NSCLC组织中都上调。LIU等报道了METTL3显著过表达[123],而另一项研究显示血清外泌体中FTO表达增加,m6A水平降低[97];(2)在相同的癌症中,m6A甲基转移酶和去甲基转移酶在治疗耐药方面表现出相似的效果。以GBM对TMZ的耐药性为例,研究证明METTL3通过增强DNA损伤修复来促进耐药性[51]。然而,FTO也被报道通过保护PDK1mRNA免于降解而增强耐药性[124];(3)对于同一类型调控因子,他们对同一疗法具有相反的调节作用。例如,METTL3和METTL14可以反向调节肝癌的索拉非尼耐药。METTL3通过抑制OATP1B1和OATP1B3表达发挥正向调控作用,降低索拉非尼摄取[79],而METTL14通过诱导OATP1B1和OATP1B3的反激活,使肝癌细胞增敏[82];(4)对于同一调控因子,对同一疗法有相反的影响。以研究最多的METTL3为代表,研究发现METTL3在NSCLC中通过刺激AKT1促进顺铂耐药[26],然而LING等人发现METTL3参与了异丙酚介导的顺铂再致敏[27]。
因此,为了澄清这些不确定性,加深对治疗耐药的认识,还需要对m6A的调控进行更多的研究。这里提供一些合理的方法,包括:(1)开发一种精确、高效的编辑工具来微调m6A对特定靶标、基因、细胞甚至特定m6A位点的修饰;(2)探索m6A调节因子在不同细胞环境、不同组织来源的癌细胞中的靶向选择性;(3)开发高选择性靶向m6A的药物,探索联合应用的可行性。
由于m6A对RNA命运的普遍调控,m6A修饰在调节肿瘤治疗耐药中发挥了重要作用,为克服耐药和优化癌症治疗奠定了基础。尽管在探索这一领域的潜在机制和临床意义方面取得了许多进展,但我们的知识还处于起步阶段,急需新的方法和技术去进行更大规模和更深入的研究。
利益冲突所有作者均声明不存在利益冲突
猜你喜欢
保健与生活(2022年11期)2022-06-09
当代水产(2021年8期)2021-11-04
生物学通报(2020年10期)2020-08-13
云南医药(2019年3期)2019-07-25
中国生殖健康(2019年12期)2019-01-07
国际妇产科学杂志(2016年2期)2016-06-16
现代检验医学杂志(2015年2期)2015-02-06
沈阳医学院学报(2014年4期)2014-12-27
遗传(2014年3期)2014-02-28
中国海洋大学学报(自然科学版)(2014年12期)2014-02-28
