
Recent research progress on small molecule compounds and its derivatives of antiparasitic drugs
2023-11-18 09:27TingWangLinWangJunHeLiChangJianyouShi
Chinese Chemical Letters 2023年10期
Ting Wang, Lin Wang, Jun He, Li Chang, Jianyou Shi
a Department of Pharmacy, Personalized Drug Therapy Key Laboratory of Sichuan Province, Sichuan Academy of Medical Sciences & Sichuan Provincial People’s Hospital, School of Medicine, University of Electronic Science and Technology of China, Chinese Academy of Sciences Sichuan Translational Medicine Research Hospital, Chengdu 610072, China
b College of Food and Bioengineering, Xihua University, Chengdu 610039, China
c Key Laboratory of Drug-Targeting and Drug Delivery System of the Education Ministry, Sichuan Engineering Laboratory for Plant-Sourced Drug and Sichuan Research Center for Drug Precision Industrial Technology, West China School of Pharmacy, Sichuan University, Chengdu 610041, China
d Emergency Intensive Care Unit, Sichuan Academy of Medical Science & Sichuan Provincial People’s Hospital, School of Medicine, University of Electronic Science and Technology of China, Chinese Academy of Sciences Sichuan Translational Medicine Research Hospital, Chengdu 610072, China
Keywords:Neglected tropical disease Parasite Anti-parasite Mechanism Crystal structure
ABSTRACT Neglected tropical diseases (NTDs) refer to infectious diseases caused by multiple pathogens that are prevalent in hot, humid climates in tropical areas.With the global economic growth and the improvement of public health status, eliminating neglected tropical diseases will be of great significance to the healthy development of human beings.However, the number of drugs and vaccines for NTDs treatment is extremely limited, so it is urgent to develop new drugs.Since most NTDs are caused by parasites, this paper selected parasitic diseases with high morbidity and mortality, and focused on new effective therapeutic targets and excellent lead compounds for these diseases.Schistosomiasis, human African trypanosomiasis (HAT), Chagas disease, leishmaniasis, filariasis and toxoplasmosis correspond to a series targets such as smHDAC8, thioredoxin glutathione reductase (TGR), T. cruzi glucokinase (TcGlcK), phosphofructokinase(PFK), type IB topoisomerase, cell division cycle-2-related Kinase, sterolmethyl transferase, calumenin, dihydrofolate reductase (DHFR) and Toxoplasma gondii farnesyl-diphosphate synthase (TgFPPs).In this paper,the pharmacological effects of typical lead compounds corresponding to each disease, the structural characteristics of the mother nucleus and the pharmacological activities of the substituent.In addition, the binding patterns of some involved targets (such as smHDAC8) with corresponding lead compounds (such as compound 13) and the signaling pathways associated with gluconeogenesis, glycolysis, and pentose phosphate pathways are analyzed in detail.In this paper, the interaction mechanism between the lead compounds and the target were thoroughly discussed, in order to provide the research ideas of potential anti-parasite compounds, and further improve the understanding and prevention ability of such diseases of NTDs.
1.Introduction
Neglected tropical diseases (NTDs) refer to infectious diseases caused by multiple pathogens that are prevalent in hot, humid climates in tropical areas [1].Up to now, World Health Organization(WHO) has divided the neglected tropical diseases into twenty categories (Fig.S1 in Supporting information), including parasitic diseases (such as schistosomiasis, leishmaniasis, onchocerciasis, soiltransmitted helminthiases, African trypanosomiasis, taeniasis, cysticerosis), bacterial infections (such as trachoma, Buruli ulcer, leprosy), viral infections (such as dengue, rabies), and spirochete infections (yaws) [2,3].Comparisons of global burden of disease data from 2000 to 2017 revealed that foodborne trematode infections,dengue and echinococcosis increased in the Asia-Pacific region by 21%, 109% and 59%, respectively [4].NTDs threaten the health of billions of people around the world, and more than 500,000 lives disappear every year [5].Data showed that the disease burden caused by NTDs is second only to human immunodeficiency virus/acquired immune deficiency syndrome (HIV/AIDS), causing economic losses of up to billions of dollars every year, and causing great economic and social burden to the countries where the disease is prevalent [6].
Although there are many types of NTDs, most of them are parasitic diseases caused by various parasites.Parasites that enter the human body migrate and multiply in the body and accumulate in tissues, organs, eyes, nerves or lymphatic systems after maturation, thereby affecting the ability of young adults to work, causing blindness or disability, hindering the development and intellectual development of children, and even causing death of patients [7].The report showed that agents of trypanosomiasis, leishmaniasis,schistosomiasis, lymphatic filariasis and onchocerciasis have caused high morbidity and mortality in countries with epidemic diseases.WHO in 2021 has even proposed a global malaria technology strategy for 2016–2030 that will reduce the morbidity and mortality of malaria cases by at least 90% [8].Therefore, this review focuses on the research progress of parasitic diseases.
The control and elimination of NTDs is one of the important tasks of public health in the world today [9].In 2021, WHO proposed to launch a roadmap for Neglected Tropical Diseases 2021–2030 to achieve the strategic goal of eliminating neglected tropical diseases.To accelerate this programmatic action, WHO has integrated NTDs into national health systems and strengthened cross-sectoral synergies to address the socioeconomic burden of NTDs [10–12].We found that in addition to improving the quality of water, strengthening case detection and management, and improving parasite-related vector control, prevention and treatment drugs are also very important.So far, although there are vaccines against dengue, schistosomiasis, leishmaniasis, Chagas disease and onchocerciasis in clinical trials, there are still few vaccines approved for clinical use [8–12].At the same time, due to insuffi-cient investment in research and development of NTDs, the number of drugs currently used to treat various parasitic diseases is very scarce (Fig.S1).Existing drugs such as artemisinin, praziquantel and ivermectin have the problem of reduced sensitivity or drug resistance [13–19].Some studies have also shown that the traditional drug named chloroquine may cause myocardial damage in patients with severe cardiotoxicity, which also reveals that some of the existing drugs have low selectivity and high toxicity [20,21].
Therefore, it is urgent to develop new drugs for the control of tropical diseases, and developing new target compounds is a promising research direction.In this paper, we reviewed the new targets for the treatment of parasitic diseases in recent years and the related signal pathways involved.We also analyzed the mother nucleus structure of typical compounds and the effects of substituents on their activities in detail.In addition, we have made a molecular docking between some targets and corresponding typical lead compounds, and analyzed the binding mode between them in detail to further understand the interaction mechanism,in order to provide research ideas for potential anti-parasite compounds.
2.Compounds associated with the treatment of tropical diseases
2.1. Schistosomiasis
Schistosomiasis is a neglected tropical disease.It is a major infectious disease that seriously endangers health and affects social and economic development, second only to malaria [13,14], and the disease is endemic in Africa, the Middle East, South America and Asia [15–17].Schistosomiasis is mainly divided intoSchistosoma intercalatum,Schistosoma mansoniandSchistosoma japonicum,which are widely distributed and seriously endangered.Acute patients are often accompanied by fever, cough, hepatosplenomegaly,chronic patients are often accompanied by fatigue, abdominal pain,intermittent diarrhea and so on [18,19].The development of new drugs is extremely difficult due to the complex life history of schistosomiasis, the changing composition of surface antigen and the interference of host immune system.Praziquantel (compound1,Fig.1) is the only drug recommended by the WHO to treat schistosomiasis in humans, however, it does not prevent reinfection and is not effective in larvae, and overuse of this drug also increases the likelihood of emergence of praziquantel resistant parasite strains [17,20,21].Therefore, the search for potential new compound targets and new drugs of antischistosome has become an urgent problem for the researchers.So far, it has been reported that a series of decoquinate derivatives have stronger activity than praziquantel [14,22].These findings have led to the development of new anti-schistosomiasis drugs.
2.1.1.Compounds acting on smHDAC8 targets
Metal dependent histone deacetylases (HDACs) are key epigenetic regulators [23].In human beings, they are one of the most studied epigenetic targets, and many HDAC inhibitors that affect cancer cells have been found [24–26].It has been proved that small-molecule HDAC inhibitors can be used to treatS.mansoni[27].Therefore, HDACs have become a potential target for the treatment of schistosomiasis.HDACs are involved in regulating gene expression, cell proliferation and cell death.SAHA (compound2), J1037 (compound3) and J1038 (compound4, Fig.1)have been confirmed as classic HDAC inhibitors in anti-cancer and anti-human parasitic diseases.SAHA was clinically approved as a chemotherapeutic drug in 2007.Compared with hsHDCA8, the class I enzyme with the lowest expression level in human beings,smHDCA8 is the most abundant class I HDAC enzyme inS.mansoni, and it is the drug target of schistosome-specific inhibitors.When the expression ofsmHDCA8 decreased, the ability of schistosome to survive and mature in host was greatly reduced [28].ThesmHDAC8 inhibitors J1037 and J1038 have poor inhibitory effect on schistosome apoptosis due to poor uptake or metabolic inactivation[29].Therefore, the design and development of effectivesmHDAC8 inhibitors has become a strategic priority.smHDAC8 is folded by HDAC with a specific external loop [30].The inverted conformation of F151 in the structure ofsmHDAC8 is highly specific for schistosomase, which made the research site ofsmHDAC8 only turn to H292 residue.The research showed that if H292 residue was replaced by polar residue, the active site will change, which also provided a direction for designing specific inhibitors targetingsmHDAC8[31–33].
Some studies have proved that some aromatic hydroxamic acid compounds weresmHDAC8 inhibitors, such as J1038, J1075 (compound5, Fig.1) and TH65 (compound6, Fig.1) and it could form hydrogen bonds with His292 residues at active sites [13,34,35].In 2018, to probe the effect of the heterocyclic group and its substitution pattern on the affinity towardssmHDAC8, Bayeret al.[36] developed different heterocyclic-hydroxamic acid derivatives.Compared to the lead compound J1075,the unsubstituted benzothiophene derivative (compound7, Fig.1) showed a 7-fold increase in thein vitroactivity towardssmHDAC8, and compound8(Fig.1) was the most potent molecule among benzothiophene derivatives.Similarly, they have also developed a series of cinnamichydroxamic acid derivatives bearing small substituents at different positions of the benzene ring among which theo–chloro derivative (compound9, Fig.1) showed the highest inhibitory potency forsmHDAC8 (IC50=60 nmol/L), but it suffered from a decrease in selectivity.Since the hydrophobic side pocket ofsmHDAC8 treated by aromatic moieties has been proved to be beneficial in terms of potency and selectivity profile,and they introduced aryloxy and arylthio moieties to improve activity and selectivity.The results showed that theortho-aryloxy/arylthio derivatives showed good selectivity for hHDAC1 and hHDAC6.It is worth mentioning that replacing the benzyloxy moiety by phenyloxy or phenylthio groups can improve the affinity of compounds, and the 4-methoxyphenyloxy derivative (compound10, Fig.1) show potent inhibition.In 2021, their group optimized the structure of compound TH65 (compound11, Fig.1), designed and synthesized a series of phenylhydroxylate derivatives as novelsmHDAC8 inhibitors,among which compound12(Fig.1) has the best potency.The crystal structure of the combination compound12andsmHDAC8 shows that the compound10binds in the active site, and the tricyclic capping group was found to be embedded in the hydrophobic side pocket ofsmHDAC8, displaying aromatic and hydrophobic interactions with H292, T341 and P291.
Based on the method of click chemistry, Shenet al.[28] found that the triazole ring may enhance the binding affinity of HDAC throughπ-πsuperposition, and obtained the triazole ring compound12with the best activity [IC50(HDAC)=104 nmol/L].Its efficacyin vitrowas equivalent to that of SAHA.In 2020, based on the optimization, Kalininet al.[37] synthesized a series of novel inhibitors of triazole ring.The results showed that phenyl derivative compound13(Fig.1) was the strongest inhibitor [IC50(smHDAC8)=4.44 μmol/L].The eutectic structure showed that compound13forms hydrogen bonds with the side chains of histidine H141 and H142 and the hydroxyl group of catalysed tyrosine Y341.The triazole ring of compound13is almost stacked between the side chains of phenylalanine F151 and F216, forming an active site tunnel (Fig.2).Because of the selectivity of compound13tosmHDAC8, they synthesized a series ofortho-substituted-1-phenyl-1H-1,2,3-triazole derivatives as new inhibitors, of which compound14(Fig.1) has the best inhibitory activity [IC50(smHDAC8)=0.504 μmol/L].The eutectic structure showed that the fluoro-phenyl capping group of compound14is stacked onto the Y341 side chain,and the triazole ring is closer to the specific residue H292 ofsmHDAC8, making compound14more selective tosmHDAC8.In summary, compound14of triazole derivatives can be developed as promising inhibitors ofsmHDAC8 in the treatment of schistosomiasis.
2.1.2.Compounds acting on TGR target
Thioredoxin glutathione reductase (TGR) is an essential enzyme for pathogens to survive in the redox environment [38,39].Because the parasite’s redox systems are completely mediated by TGR enzyme, TGR has become a potential drug target for the treatment of schistosomiasis.There are three enzyme activities including thioredoxin reductase (TrxR), glutathione reductase (GR) and glutathione reductase (Grx) [40,41].It was found that the redox activity of enzymes depends on at least three redox sites that communicate with each other: the first was the FAD site composed of the isoalloxazine ring of the flavin and Cys154-Cys159 pair, the second was the C terminal composed of Gly-Cys-Ser-Gly-sequence, and the third was the glutaredoxin redox site composed of Cys28-Cys31 at N terminal [42].It was found thatSchistosomawas exposed to reactive oxygen species (ROS) through respiration and host immune response.For the first time, the research team found thatS.mansonihas an unusual thiol redox system that integrates thioredoxin and glutathione into an enzyme TGR to minimize oxidative stress,thereby protecting the host from pathogeninduced oxidative stress[43,44].In addition, RNA interference with TGR expression assay showed that the parasite died in a short period of time, further confirming that TGR is essential for parasite survival.

Fig.2.Binding mode of compound 13 with smHDAC8 (PDB: 6TLD).(A) The overall structure of smHDAC8.(B) The binding mode of compound 13 and smHDAC8.Hydrogen bonds are shown in green dash.This figure was drawn by PyMOL software (https://www.pymol.org).
Angelucciet al.[44] found that auranofin (compound15, Fig.1)used to treat rheumatoid arthritis could show antischistosomiasis activity by inhibiting TGR enzyme.Consistent with previous studies, compound15could partially release Au atom to protein thiols and free thiols.Through ligand replacement reaction, Au atom was transferred from serum albumin to other proteins or thiols in the body until reaching the receptor and eventually transferring Au atoms to the active Cys-pair of the TGR enzyme [44–46].Sayedet al.[47] found that oxadiazole-2-oxide could effectively inhibit TGR and cause parasite death through high-throughput screening and confirmatory experiments.The most effective compound16(Fig.1), even showed superior inhibitory activity to the control drug praziquantel.Oxadiazole-2-oxide has lower cytotoxicity,higher activity and tolerance in mice, which provided a direction for the design and research of new small molecule drugs against human schistosomiasis.Peroxiredoxin (Prx) is one of the major antioxidant enzyme families of parasites, which a peroxidase superfamily widely present in prokaryotic and eukaryotic organisms,and plays an important role in the antioxidant effect of organisms.Simeonovet al.[48] screened the inhibitory effect of TGR/Prx in the form of double enzymes based on previous studies on TGR.They carried out automatic quantitative high-throughput experiments on a series of compounds collected in the previous period, and identified three series of active compounds with IC50values ranging from micromolar concentration to the determination response limit of 25 nmol/L were identified with certain inhibitory effects on TGR.This was the first report of large-scale high throughput screening (HTS) identifying lead compounds for worm diseases, and provided an example for initiating the development of new treatments for other neglected tropical diseases.Raiet al.[49] continued to study a series of oxadiazole-2-oxide compounds and found that TGR was a dehydrogenase containing selenocysteine, which was required by parasites to maintain the redox balance of cells.Through the systematic evaluation of the core structure of oxadiazole-2-oxide compounds, they found that removing the nitrogen-oxygen dipole or changing the position of the nitrogen-oxygen dipole will lose the inhibitory activity and insecticidal effect of TGR.Therefore, it was inferred that the nitrogen and oxygen dipoles are the pharmacodynamic group of the compound, which proved that oxadiazole-2-oxide could be used as a new type of TGR inhibitor.In 2015, Johannet al.[50] found a series of 2-methyl-1,4-naphthoquinone derivatives and evaluated theirSmTGR and anti schistosome inhibition activity.Among them,3-phenoxymethylenedione has high potential to selectively inhibitSmTGR.Compounds17and18(Fig.1) were identified as the most potentSmTGR inhibitors.It was found that compound17showed a time-dependent inactivation ofSmTGR, which was related to unproductive nicotinamide adenine dinucleotide phosphate (NADPH)-dependent redox cycling ofSmTGR.At the same time, compound17also showed a potent anti-schistosome action in worms culturedex vivo.Compound18inactivatedSmTGR through an irreversible non-consuming NADPH-dependent process, and has almost no killing effect on wormsin vitro.This indicated that improving the pharmacokinetic properties and bioavailability is a desirable research direction for future TGR inhibitors.
2.2. Human African trypanosomiasis and Chagas disease
Trypanosoma bruceiis the pathogen of human African trypanosomiasis (HAT) [51].After infection, parasites can penetrate the blood barrier and invade the central nervous system through the lymphatic system, causing a series of serious neurological manifestations, even coma and death [52].Drugs currently used for the treatment of HAT are generally not available for all cycles of growth and are poorly selective, leading to frequent side effects,and pentamidine (compound19, Fig.3A) could not be used for HAT second-stage treatment, which made the treatment method have significant defects [53].Trypanosoma cruzicauses Chagas disease, an acute phase characterized by skin damage, fever and dyspnea that may threaten the life of patients with weakened immune systems, and a chronic phase characterized by organ inflammation that can lead to cardiomyopathy, cardiac arrest and heart failure[54].At present, the available anti-phagocytotic drugs only include benznidazole (compound20, Fig.3A) and nifurtimox (compound21, Fig.3A) [55].However, when these two drugs are administered in the chronic phase, the therapeutic effect is significantly reduced,and may have side effects such as severe peripheral neuropathy,vomiting, nausea and insomnia [56].Without proper medical care,Chagas disease andLeishmandisease was also fatal, coupled with the increasingly serious drug resistance, so it was urgent to find new therapeutic drugs to combat this disease.It has been reported that compounds with 4-aminoquinoline groups exhibit insecticidal and antimalarial activities [57].
It has been reported that compounds with 4-aminoquinoline groups exhibit invasive and antimalarial activities [57].In 2015,Pietroet al.[46] found that the tricyclic skeleton of the previously synthesized acetylcholinesterase (AChE) inhibitor (compound22, Fig.3A) contained 4-aminoquinoline, and speculated that it may have certain antigenicity.Compound23(Fig.3A)showed anti-biological activity againstT.bruceiwith an IC50value of 3.3 μmol/L through experimental verification.On the basis of compound23, they took compound22as the structural basis and multi-component reaction as the reaction condition to carry out biological analysis on it, and they designed and synthesized novel benzo[h][1,6]naphthyridines, pyrrolo[3,2-c]quinolines, azepino[3,2-c]quinolines, and pyrano[3,2-c]quinolines derivatives with anti-trypanosoma potency.Several tricyclic heterofused quinoline derivatives were found to display an interesting multi-trypanosomatid profile, among them, compound23has a high inhibitory effect onT.brucei,L.infantumandT.cruziwith IC50values of 1.5, 6.1 and 29.2 μmol/L, respectively.It was also showed a higher expected score of MPO of the central nervous system, better pharmacokinetic properties and lower P-gp efflux.Compared with other derivatives, it showed the lowest activity of acetylcholinesterase and became the optimal structure.When exploring the structure–activity relationship of these compounds,they found that the bromine substituent at position 1 of ring A had the best inhibitory activity, while -NH substituent had the weakest activity.The anti-brucella activity ofN-substituted derivatives of ring-opening quinoline analogues were stronger than that of -OH and cyano-substituted derivatives (Fig.3B).In addition, it was also found that the presence of the oxidized B ring with protonable 4-(aminomethyl) phenyl group could be more effective againstT.bruceiandT.cruzi.Interestingly, all these multitrypanosomatid compounds were predicted to span blood brain barrier, which is crucial for the treatment of advanced HAT.In addition, these compounds have lower lipophilicity and better pharmaceutical properties than compound22.In conclusion, pyranoquinoline compound23was a good lead compound against trypanosomes.In the follow-up design, the design should also focus on reducing acetylcholinesterase inhibitory activity and increasing the selectivity index of drugs to reduce drug side effects and increase efficacy.

Fig.3.(A) Chemical structures of compounds 19–30.(B) The structure–activity relationship of pyranoquinoline derivatives.(C) The structure–activity relationship of p-aminosulfonamide derivatives.
2.2.1.Compounds acting on TcGlcK target
TcGlcK, a TP-dependent glucokinase, is an important metabolic enzyme inT.cruzi.In the presence of Mg2+and ATP,T.cruziglucokinase phosphorylates glucose into glucose-6-phosphate (G6P), a key intermediate in glycolysis, gluconeogenesis and pentose phosphate pathways.G6P is critical to the production of TP in parasites and the formation of NADPH and biosynthetic precursors through the pentose phosphate pathway, and plays an important role in maintaining cellular antioxidant activity and nucleotide/nucleic acid synthesis [58–60].By inhibiting glucokinase,TcGlcK inhibitors restrict the conversion of D-glucose to G6P (Fig.4).In order to maintain energy supply and meet the requirements of biosynthesis, the gluconeogenic pathway ofTrypan cruzansuses more ATP, GTP and NADH from glycosomal reserves [60].Based on the above reasons, the depletion of the nucleotide and electron carrier reserves results in the death of the Cruze trypanosomes.In 2007, Cordeiroet al.[58] presented a complete eutectic structure ofTcGlcK combined with substrate (β-D-glucose) and its reaction products.The crystal structure ofTcGlcK contained a homodimer in the asymmetric unit.Theβ-D-glucose and ADP molecules bound to eachTcGlcK monomer at the active site between these large and small domains.The substrateβ-D-glucose bound to residues Asn105, Asn130, Asp131, Glu207 and Glu236 through hydrogen bonds.The adenine N1 part of ADP interacts with Asn312 through bicentate hydrogen bonds, while the N3 part interacts with the carbonyl group of Gly184 and the hydroxyl group of Tyr-through water molecules.In addition, the ribose portion of ADP interacts with Asn308 and Thr185.
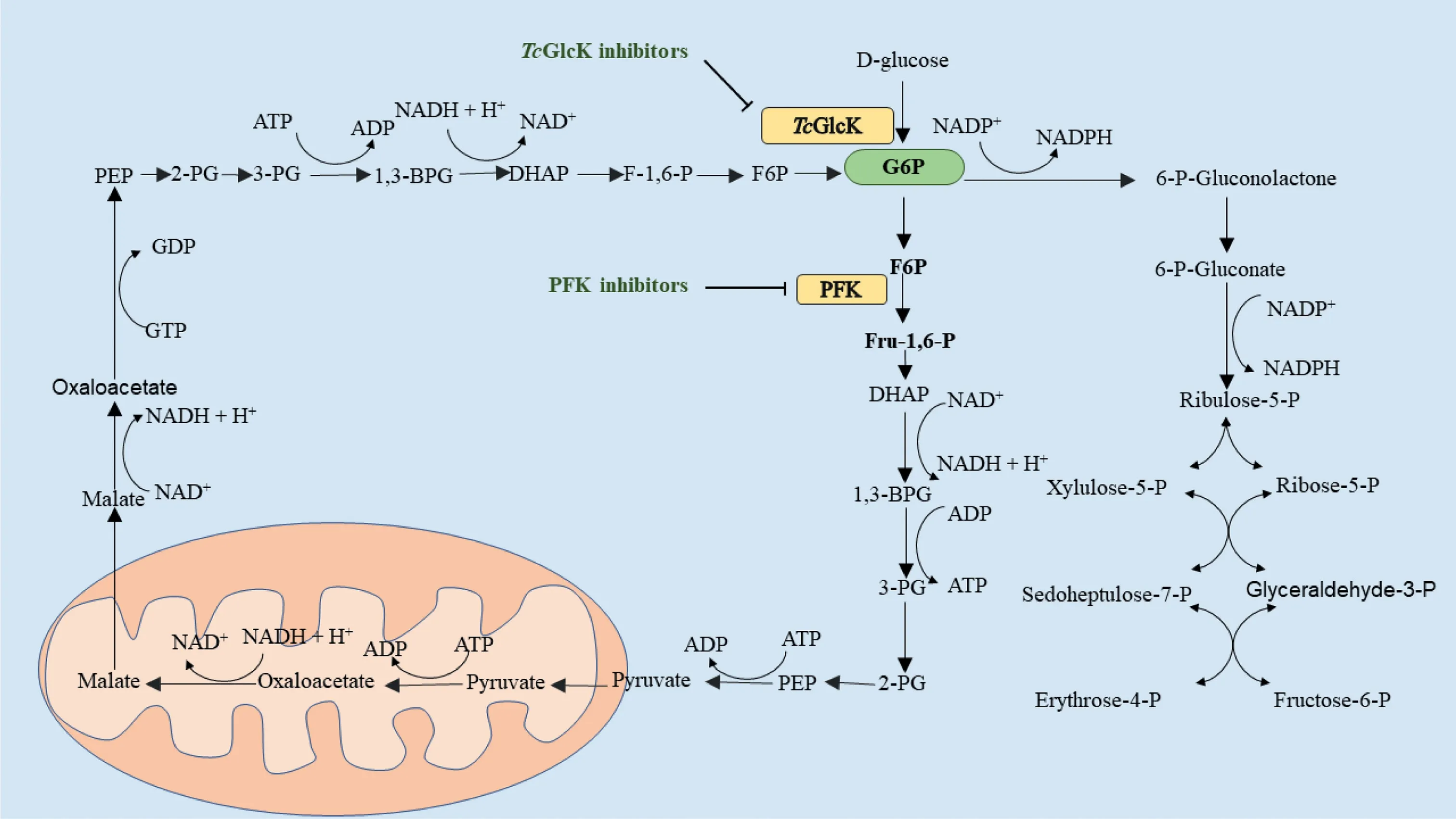
Fig.4.Gluconeogenesis, glycolysis and pentose phosphate pathway in Trypanosoma cruzi.PEP, phosphoenolpyruvate; 2-PG, 2-phosphoglycerate; 1,3-BPG, 1,3-bisphosphoglycerate; DHAP, dihydroxyacetone phosphate; F-1,6-P, fructose 1,6-bisphosphate; F6P, fructose-6-phosphate; G6P, glucose-6-phosphate; 6-P-Gluconolactone,6-phosphogluconolactone; 6-P-Gluconate, 6-phosphogluconate; Ribulose-5-P, ribulo-5-phosphate; Xylulose-5-P, xylulose-5-phosphate; Ribose-5-P, ribose-5-phosphate;Sedoheptulose-7-P, sedoheptulose-7-phosphate; Glyceraldehyde-3-P, glyceraldehyde-3-phosphate; Erythrose-4-P, erythrose-4- phosphate; Fructose-6-P, fructose-6-phosphate.
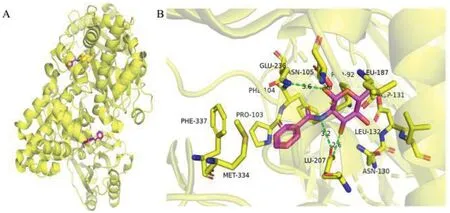
Fig.5.Binding mode of Benz GlcN with TcGlcK (PDB: 5BRD).(A) The overall structure of TcGlcK.(B) The binding mode of Benz GlcN.This figure was drawn by PyMOL software (https://www.pymol.org).
Structure based drug design (SBDD) is a drug design method based on molecular recognition based on the premise of threedimensional structure of biological targets, which has become one of the important tools for drug discovery [61–63].In 2015,D’Antonioet al.[64] designed and synthesized four glucose analogues with inhibition ofTcGlcK activity based on SBDD and Xray crystallography methods and the idea of competitive inhibition.It was found that compound24(CBZ-GlcN, Fig.3A) and compound25(Benz-GlcN, Fig.3A) showed good bioactivity againstT.cruzi in vitro, and Benz-GlcN showed the strongest inhibitory effect on glucokinase with theκivalue of 0.71 μmol/L.X-ray crystallography showed that the phenyl tail in theTcGlcK inhibitor structure interacted with F337, P103 and N105 side chains (Fig.5).When the tail group of the inhibitor was an aromatic group,the inhibitor would formπ-stacking interaction with the F337 residue of the dimer.The results showed that there was an important hydrophobic capsule surrounding the glucose binding site ofTcGlcK, representing the affinity and selectivity ofTcGlcK glucoamine analogue inhibitors.Based on previous research, Greenet al.used compound25as a lead compound to design novel libraries of amino sugar analogues containing D-glucosamine (DGLCN), D-mannosamine (D-Mann), or D-galactosamine (D-GalN)skeletons in 2021 [65].It was found that five compounds containing D-GlcN skeleton showed good inhibitory activity, most of the compounds were strongTcGlcK inhibitors because theirκivalue did not exceed 20 μmol/L.Compound25became an excellent lead compound againstC.cruziawith the best inhibitory activity(IC50=16.08±0.16 μmol/L).Therefore, the development of amino sugar analogs of D-GLCN skeleton is expected to find more effectiveTcGlcK inhibitors for the treatment of HAT and Chagas disease.
2.2.2.Compounds acting on PFK target
Phosphofructokinase (PFK), also known as 6-phosphofructokinase 1, can promote the conversion of fructose-6-phosphate (F6P) to fructose-1,6-diphosphate.Since the enzyme is inhibited by high concentration of ATP, its binding curve with substrate F6P changes from S-type to hyperbolic type, thus slowing down process of glycolysis and is the key rate-limiting enzyme of glycolysis (Fig.4) [66,67].In addition, the enzyme can also indirectly catalyze PEP into pyruvate to affect the glycolysis pathway.RNA interference (RNAi) experiments also showed that the survival rate of cultured parasites could be significantly reduced by only partially reducing the overall glycolytic flux [68].In addition, the highly inimitable characteristics ofTb-PFK make it highly selective and harmless to human body [67].
Thep-aminosulfonamide compound26(ML251, Fig.3A) was the first compound with submicromolar inhibitory activity againstTrypanosome bruceiphosphofructokinase, showed good activity in parasite growth assays (IC50=370 nmol/L, ED50=16.3 μmol/L) and did not inhibit the bacterial subtype of PFK with good selectivity[69–71].Walshet al.[72] identified a group of active PFK inhibitors by high-throughput screening with compound26(Fig.3A) as the lead compound, among which compound27(Fig.3A) with an IC50values of 410 nmol/L showed ideal activity curve andin vitrometabolic characteristics.In the study of the optimized structure,they found that 1,4-disubstitutedp-aminosulfonamide benzene ring was the best scaffold (such as compound28, Fig.3A), benzylmethylene was an important group to maintain activity (such as compound29, Fig.3A), and compounds with lipophilic 3,4-dimethoxybenzyl substituents had good tolerance (such as compound30, Fig.3A).Therefore, the structure-activity relationship of this kind of compounds were shown in Fig.3C.The optimal substituents of R1 and R2 were 3,4-dichlorophenyl and five-membered heterocyclic (isoxazole), respectively.In conclusion, compound26and its derivatives were excellent lead compounds acting on PFK targets, which could be used in the subsequent development of anti-trypanosomiasis.

Fig.6.Chemical structures of compounds 31–42.
2.3. Leishmaniasis
Leishmaniabelongs to the trypanosomatidae family [73].Its life stages include two periods: promastigote and amastigote.The promastigote parasitizes in the digestive tract of the vector insects(Chrysopidae), and the amastigote parasitizes in the megaphagocytes of mammals or reptiles [74,75].Leishmaniainfects humans through chrysopidae as a transmission medium, causing leishmaniasis, also known as black fever.Its clinical manifestations are long-term irregular fever, hepatosplenomegaly, anemia,etc.[76].Due to the huge burden of leishmaniasis and the shortcomings of high toxicity, drug resistance and inability to cure the disease [77],leishmaniasis has been included as one of the tropical diseases under enhanced control by the WHO, which urgently needs to find new targets.
Rajet al.[78] discussed severalLeishmaniametabolic pathways,including sterol biosynthetic pathway, glycolytic pathway, and DNA topoisomerases, which might be exploited for the development of effective antileishmaniasis strategies.In 2016, Khareet al.[79] found that GNF6702 (compound31, Fig.6) containing triazole pyrimidine skeleton, a selective inhibitor of the proteasome of motids, showed potent activity againstLeishmaniabecause of its role in inhibiting the proteasome protein degradation mechanism of cells [EC50(L.donovani)=18 nmol/L].In 2021, Santiago-Silvaet al.[80] found that benzoylguanidines could be a starting scaffold for the search for new antileishmanial drugs.Compounds32and33were found to cause mitochondrial depolarization and increase ROS levels, and to generate autophagy vacuoles in free promastigotes.They could also inhibit the proliferation ofLeishmaniaby increasing induction of microbicide molecule NO.Currently, the drugs used to treat the disease mainly include polyene antifungal drugs amphotericin B (compound34, Fig.6),miltefosine (compound35, Fig.6) and sodium stibogluconate(compound36, Fig.6) [81,82].It is worth mentioning that in 2022,Seifuet al.[83] discovered a group of quinazolinones derivatives with poteny antileishmania activity.Among them, compound37was the best structure (IC50=0.0212 μg/mL), and it was 2 and 150 times more active than amphotericin B and miltefosine,respectively.
2.3.1.Compounds acting on type IB topoisomerase target
DNA topoisomerase is an important enzyme in the nucleus,which can catalyze the cleavage and binding of DNA strands.They are categorized into two subclasses as follows: type I topoisomerase which unwinds the single-stranded DNA and type II topoisomerase that cleaves the double-stranded DNA [82,84,85].Type IB topoisomerase can release the torque accumulated in the DNA chain, and consume topoisomerase by preventing the enzyme from binding to the substrate DNA or inhibiting the formation of covalent cleavage complex, so as to control the topological state of DNA, thus affecting cell processes such as replication and transcription, and affecting cell proliferation [82,86].Since DNA topoisomerase has a heterodimeric architecture different from human topoisomerase I, it has been confirmed that type IB topoisomerase was an excellent target for inhibitingLeishmaniaandTrypanosoma Brunei[87].So far, only a few natural products, such as dihydrobetulinic acid (DHBA, compound38, Fig.6) and dihydrobetulin derivatives have been reported [88], and the catalytic inhibitors ofLdTopIB have not been studied in depth [89].

Fig.7.Chemical structures of compounds 43–59.
In view of the high medicinal value of spirooxindole like natural products, such as insecticidal, anti-tumor and antibacterial [90–93].Rottmannet al.[94] found that spirocyclodone NITD609 (compound39, Fig.6) is an effective candidate drug for malaria.Scalaet al.[95] further found that C3-mono-functional oxindoles and spirooxindoles (compounds40and41, Fig.6) could inhibit the proliferation of pro flagella and sterile flagella-freeLeishmaniain a dose-dependent manner, so spirooxindoles could be considered as effective anti-Leishmania drugs for development.By screening spirooxindoles and bis-spirooxindoles derivatives, Sahaet al.found only compound42(Fig.6) could kill wild type AG83 strain ofL.donovaniand resistant strains GE1, MIL-R and CPT-R by inhibitingLdTopIB pathway, and had less toxicity to host macrophages.Job Plot analysis established that two molecules of compound42is binding to one molecule ofLdTopIB.It means that there was a 2:1 interaction of this inhibitor withLdTopIB.Molecular docking of compound42withLdTopIB showed that two binding sites on bisubunit enzymeLdTopIB.Compound42may have binding sites on small subunit and hinge region of large subunit ofLdTopIB.In addition, Leu-215, Gly-439, Lys-219, Ile-220, Ile-228, Lys-241, Ile-242,Phe-243 and Ile-247 of the small subunit were found to be the most important amino acids in the interaction with compound42.Leu-437, Phe-441, Asn-441 and Asn-444 of large subunit were also involved during this binding to small subunit ofLdTopIB.Therefore,potentially compound42can be used as a lead compound for developing excellent anileishmanial agent against emerging drug resistant strains of the parasite.
2.3.2.Compounds acting on CRK12 target
Cell division cycle-2-related kinase (CRK3/CRK6/CRK12), a cyclin-dependent kinase (CDK), is an essential gene forLeishmania Donovaniand plays a key role inTrypanosoma bruceiblood flow phase [96].The lack of CRK12 will lead to flagellar expansion and impaired endocytosis, which directly affects the survival of parasites [97–100].In 2018, Wyllieet al.[97] reported that the main mechanism of the pyrazolopyrimidine compound they designed is to treat leishmaniasis by inhibiting CRK12, and GSK3186899(compound43, Fig.7) is currently under pre-clinical development(EC50(Amastigotes)=0.017 μmol/L).Therefore, CRK12 was defined as one of very few chemically validated drug targets inLeishmania[97].
In 2021, Broniet al.[101] modelled the 3D molecular structure of theL.donovaniCRK12 (LdCRK12) and screened for small molecules with potential inhibitory activity from African flora.Among them, the inhibition constantκiof the natural compound sesamin NANPDB1649 (compound44, Fig.7), methyl ellagic acid NANPDB1406 (compound45, Fig.7), styrene NANPDB2581 (compound46, Fig.7) and sensenoside NANPDB6446 (compound47,Fig.7) was between 0.108–0.587, which were found to be a potentialLdCRK12 inhibitor.The molecular docking studies revealed that the binding affinities of compounds42and43againstLdCRK12 were higher than those of the known inhibitors and drugs, including GSK3186899, amphotericin B and miltefosine.In addition to the study ofin vitroinhibitory activity, suitable physical and chemical properties are also very important factors in new drug development.When the LE-dependent lipophilicity (LELP) value is between 0 and 7.5, the compound can activate specific signaling pathways through the blood-brain barrier to achieve ideal efficacy.It was found that the LELP values of compounds45and47were 3.761 and 0.521, respectively, which had good physical and chemical properties and could be used as ideal lead compounds for further study.Interestingly, the team also found that Lys488, which was bound to the ATP-binding pocket of the kinase domain, was a very critical amino acid residue for ligand binding in the ATPbinding site.Therefore, the potential antileishmanial compounds could serve as templates for fragment-based drug design forLeishmaniainhibitors.
2.3.3.Compounds acting on TryR target
Oxidative stress plays a crucial role in the host’s immune fight against infection [102–104].To a large extent, the ability to resist this oxidative attack is related to the survival of parasites.Due to the lack of hydrogen peroxide enzyme and some traditional redox control systems inLeishmaniaand infectiousTrypanosoma[105],they maintain the balance of thiol internal environment through an unusual variant of glutathione-trypanothione [106].Maintaining this trypanothione in a reduced state requires the involvement of trypanothione disulfide reductase (TryR), which is essential forTrypanosoma’s antioxidant defense.TryR is a homodimer with two separate subunits, including two active sites, FAD domain and NADPH binding domain.FAD cofactors restore cysteine by transferring two electrons from NADPH to Cys52 and Cys57,while oxidized TS2binds to protein, and then Cys52 attacks the disulfide bond of substrate to release reduced T(SH)2.Since TryR only exists in parasites and plays a key role in parasite survival,TryR has become a target for the study of resistance to leishmaniasis, and the study of novel TryR inhibitors has also become a research direction [107–109].Revueltoet al.[110] reported that inhibition ofLeishmania infantumTryR (LiTryR) by disruption of its homodimeric interface is an alternative strategy in the search for novel antileishmanial agents.In this study, they found and verified that E426 was the key amino acid affecting the structural stability and function of TryR dimer.On this basis, the structural scaffold obtained by theα-helix method could be used as dimerization interference of TryR was published in 2021.In this library,the team selected a series of triazole-phenyl-thiazole compounds for research, and they found that compounds48–51(Fig.7) with the IC50values of 5.9, 10.9, 4.3 and 0.4 μmol/L could be used as TryR inhibitors for subsequent studies.It is worth mentioning that compound51emerged not only as the most effectiveLiTryR inhibitor of the entire series, but also as a very potent dimerization disruptor.The structure-activity relationship of this kind of compounds pointed out that the presence of a positively charged amino alkyl group was an absolute requirement for inhibitory activity and dimerization, which laid the foundation for innovative TryR inhibitors.
2.3.4.Compounds acting on sterolmethyl transferase target
Ergosterol inLeishmaniaparasite cells is necessary for parasite survival,Δ24-sterolmethyl transferase (SMT) is one of the key enzymes affecting ergosterol synthesis.SMT inhibitors only affect parasitic cells without damaging higher eukaryotes because there is no such enzyme in human cholesterol biosynthesis pathway,which have high selectivity and can greatly reduce clinical adverse reactions [111–113].
The research direction of SMT inhibitors is glucocorticosteroids with heteroatoms (N or S) to replace C-24 or C-25 in order to simulate carboncation intermediates and thus bind tightly to enzymes[113].Metal-containing drugs have been widely used as antitumor drugs in recent years, but few studies have used them as antiparasitic drugs [114].It has been demonstrated that linking chloroquine or clotrimazole to metal-containing fragments can enhance biological activity against malaria and Chagas disease, respectively[115].Visbalet al.[116] synthesized two newtrans[Pt(Hpy1)2(Cl)2](compound52, Fig.7) andtrans[Pt(Hpy2)2(Cl)2] complexes (compound53, Fig.7) in order to againstLeishmania.These two complexes were specific inhibitors of SMT, which could cause serious loss of movement, swelling, vacuolization and death of parasites.Further studies revealed that Hpy1 produced higherLeishmaniainhibitory activity than Hpy2 (39%vs.16%).It was the first report on the use of Pt coordinated sterol hydrazone as a new anti-parasitic chemotherapeutic that could be further investigated as a lead compound for a new SMT inhibitor againstLeishmania.
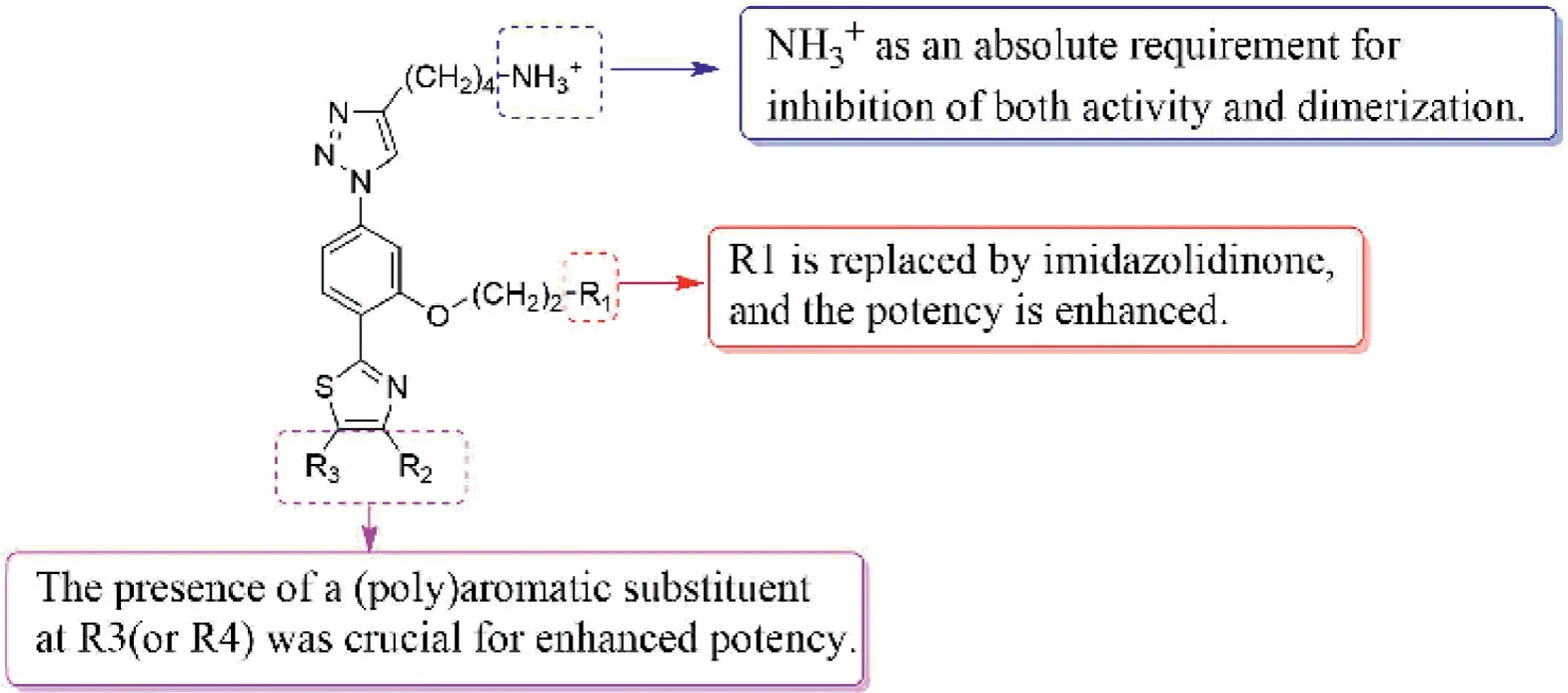
Fig.8.The structure–activity relationship of triazole-phenyl-thiazole derivatives.
2.3.5.Compounds acting on other target
Since anticancer drugs can affect DNA metabolism, protein kinase pathway, glucose catabolism, it has been reported that some anticancer drugs have the potential to killLeishmania in vitro[117–119].It is speculated that these can be used as effective drugs for the treatment ofLeishmania[120].Since the growth of parasites in the host and the growth of cancer cells exhibit two common characteristics including the ability of rapid cell division and the existence of immune evasion and defense [121].At the same time,it has also been reported that several protozoon and unicellular eukaryotes, includingLeishmania, showed molecular mechanisms of programmed death with similarities to apoptotic mechanisms of cell suicide in multicellular organisms, including cytochrome C release and DNA fragmentation [122].It has been confirmed that the standard anticancer drugs oxaliplatin and 5-fluorouracil exhibit significant antileishmania activityin vitro, which can be prospectively used as a good source of lead compounds against leishmaniasis and other parasitic diseases [123,124].Sandersonet al.[125] found that sunitinib (compound54, Fig.7), sorafenib (compound55, Fig.7) and lapatinib (compound56,Fig.7), the protein kinase inhibitors used for the treatment of cancer, had significant aboriginal activities against the amastigotes ofLeishmania donovaniwith IC50values of 1.16, 4.23 and 3.27 μmol/L, respectively.It showed similar efficacy to the positive control drug miltefosine and were not cytotoxic to mammals.
Heterocyclic compounds have been reported as the mother nucleus for anti-leishmaniasis research [126].In 2017, Masoodet al.[127] designed a series of 1,2,3-triazole-amino acid derivatives as new anti-leishmaniasis drugs by introducing triazole efficacy groups and hydrophobic substituents into amino acids with the starting point of compound hydrolysis resistance.With miltefosine as a positive control, a series of synthesized compounds were tested for anti-leishmaniasis activityin vitroand cytotoxicity towards THP-1 macrophages.Research on the structure–activity relationship of 1,2,3-triazole-amino acid derivatives found the activity of -CH3substituent compounds containing phenylalanine tail were the best, the activity of substituent-free compounds containing proline tails were still preserved, the activity ofp-OCH3substituent compounds containing tryptophan tails were enhanced (Fig.8).Compounds57–59(Fig.7) showed good activity againstLeishmaniawith the IC50values of 88.83, 96.88 and 94.45 μmol/L, respectively.Compound57also showed the lowest cytotoxicity with the IC50values of 780.01 μmol/L, which was 14 fold lower than that of miltefosine.In conclusion, compound57with the 1,2,3-triazole-amino skeleton could be further studied as a lead compound againstLeishmania.
2.4. Filariasis
Fig.9.Chemical structures of compounds 60–71.
Lymphatic filariasis and onchocerciasis have a considerable incidence in tropical countries [128,129].Chronic infection often leads to lymphedema or thickening of skin tissue in the extremities and can lead to eye damage or even blindness [130].According to WHO, hundreds of millions of people are now infected and more than 40 million are disfigured or disabled by this disease [129,131].Currently, ivermectin (IVM, compound60, Fig.9), diethylcarbamazine (DEC, compound61, Fig.9) and albendazole (ALB, compound62, Fig.9) are commonly used in clinical practice.However, it is less effective in killing imago of filariasis [132–135].At present, there is still no suitable drug for clinical use, so it is urgent to develop new target drugs.
2.4.1.Compounds acting on calumenin target
Calumenin, a calcium-binding protein, is located in the endoplasmic network cavity, where proline 4-hydroxylation occurs, and is involved in the reproduction and cuticle development of filarial nematodes, which is a key protein in normal cuticle development[136].In subsequent studies, it was found that the dysfunction of the cuticlar may directly lead to the failure of effective drug delivery in the worm.In addition, the loss of calglutenin function has also been confirmed to reduce the fertility of filariasis [137].Therefore, it can be identified as a specific drug target for filariasis.Since the safety properties of marketed drugs are guaranteed, there is less risk of re-adjustment drugs, and drugs with known safety and pharmacokinetic characteristics can be discovered more quickly[138].The research team [132] used predictive protein models to perform virtual screening in a library of marketed drugs and identified several candidate drugs such as daunomycin hydrochloride,idarubicin, paliperidone and itraconazole (ITC, compound63, Fig.9) that could react specifically with calumenin.However, the target selectivity of these three compounds between parasites and hosts was not ideal, so they were excluded from further studies.At the same time, the molecular docking results showed that the binding of ITC to calumenin was similar to that of lanosterol 14-α-demethylase and ITC.The triazole group of ITC was docked with the binding capsule of calumenin and interacts with residues of Lys164, Asn165, Glu174 and Asp161.At the same time, the long tail of ITC filled the entry channel of the two molecules.However, the effective concentration of ITC was much higher than the known anti filariasis drugs (ALB and IVM).The structure of ITC must be further optimized on this basis to develop more effective and safe specific calumenin inhibitors.
2.4.2.Compounds acting on dihydrofolate reductase target
Dihydrofolate reductase (DHFR), the sole source of tetrahydrofolate, is one of the key enzymes in the folate metabolism pathway and a suitable target for antibacterial, anticancer and antiparasitic therapies [139,140].Molecular biological evaluationin vitroand folic acid reversal studies showed that folic acid pathway plays an important role in antifiliasis drugs [141].At present, DHFR antagonists used in clinical include trimethoprin (compound64, Fig.9)and pyrimethamine (compound65, Fig.9), which have problems of low efficiency and drug resistance [142–145].Baget al.[146] found that biguanide and dihydrotriazine derivatives had inhibitory activities against Malayan filariasis, which were superior to trimethoprim and pyrimethamine.On the basis of previous studies, Sharmaet al.[147] found that compounds with 2,4-diaminopyrimidine and 2,4-diamino-striazines scaffold could also inhibit DHFR targets and effectively inhibit filamentous worms including microfilariae and adults.The effects of these drugs are permanent and irreversible.It was found that excellent compounds66,67and68(Fig.9) could completely lose the motility of parasites, and their effects were also better than those of conventional drugs such as trimethoprim and pyrimidine.The mechanism of action showed that these compounds could make the standard inducer of apoptosis of botrycin show fluorescence, indicating the occurrence of apoptosis.As PARP is a key enzyme involved in DNA repair, in the process of apoptosis, cysteine proteinase-3 cleavage of PARP leads to the reduction of PARP enzyme activity, thereby preventing apoptotic cells from repairing their own DNA, so inhibition of PARP activity is an important means to evaluate apoptosis [148,149].Compared with the negative control, the inhibition rates of these three compounds on PARP activity were 61.42%, 73.58% and 51.18%, respectively.In conclusion, compounds66,67and68can be used as outstanding lead compounds for the subsequent optimization of DHFR inhibitors in the treatment of filariasis through apoptosis.
2.5. Toxoplasmosis

Fig.10.Schematic of isoprenoid metabolism through the non-mevalonate pathway.GAP, glyceraldehyde 3-phosphate; Pyr, pyruvate; DSX, 1-deoxy-D-xylulose 5-phosphate synthase; DOXP, 1-deoxy-D-xylulose 5-phosphate; DXP-RI, DXP reductoisomerase; MEP, 2-C-methyl-D-erythritol; CDP-ME, CDP-2-C-methyl-D-erythritol; cMEPP, 2-C-methyl-D-erythritol-2,4-cyclodiphosphate; DMPP, dimethylallyl diphosphate; IPP, isopentenyl diphosphate; GPP, geranyl diphosphate; FPP, farnasyl diphosphate; GGPP, geranylgeranyl diphosphate.
Toxoplasma gondii(T.gondii) is one of the most common parasites in the world [150].Toxoplasmosis caused byToxoplasma gondiiinfection is a common zoonosis and one of the neglected tropical diseases listed by WHO [151].Epidemiological data showed that about 30%–50% of the global population has been infected with toxoplasmosis, and 10%–20% of acutely infected patients have lymphadenectasis or flu-like symptoms.Toxoplasmosis in immunodeficient patients often leads to neurological diseases[152].Some data even show that the infection rate ofT.gondiiin tumor patients is significantly higher than that in non-tumor patients [153–155].The current drugs used to treat toxoplasmosis have some disadvantages, such as high price, high side effects and insufficient access to the central nervous system [156,157].There is an urgent need for safe and effective new toxoplasmosis treatments.Isoprenoids play an important role in all organisms and are essential compounds [158].Isoprenoids inT.gondiiare derived from isopentenyl diphosphate (IPP) and its isomer dimethylallyl diphosphate (DMAPP) generated through 1-deoxy-D-xylose-5-phosphate (DOXP) pathway (Fig.10), whileToxoplasma gondiifarnesyl-diphosphate synthase (TgF-PPS) can catalyze the formation of FPP, so it is speculated that acting on the target ofTgFPPS can inhibit the occurrence of toxoplasmosis [159].Urbinaet al.[160] found for the first time that drugs commonly used in clinical treatment and prevention of osteoporosis, such as pamidronate(compound69, Fig.9) and alendronate (compound70, Fig.9),could effectively inhibit the growth ofTrypanosoma cruzi,Toxoplasma gondii,LeishmaniaandPlasmodium falciparumwithout toxicity to host cells.The use of these marketed drugs could be expanded if they are applied to the clinical treatment of parasitic diseases [161,162].Further studies revealed [163] thatT.gondiiFPPS were the main molecular targets of bisphosphonates, so bisphosphonates could effectively inhibit the growth of the parasite.On the basis of previous studies, Szajnmanet al.[164] found that linear diphosphonates were more effective than amino diphosphonates in inhibiting parasites.The most potent compound (compound71, Fig.9) is a sulfone-containing compound, which had an EC50of 0.11 μmol/L against intracellular tachyzoites.Compound71showed low toxicity when tested in tissue culture, it also showed high activity in a toxoplasmosis mouse model.In conclusion, the inhibitory effect of this sulfone bisphosphonate derivative reported for the first time by the research team onT.gondiiwas significantly improved compared with the reported compounds, which can be used as an efficient lead compound for subsequent research.
3.Perspective
NTDs are a group of disease syndroms caused by multiple pathogens.After human beings are infected, the parasites migrate and multiply in the body quickly.When they mature, they are easy to accumulate in human tissues, organs, eyes, nerves or lymphatic systems, threatening the health of billions of people worldwide.However, there are few drugs used in clinical treatment of parasitic diseases, and some drugs have low inhibitory activity.Some drugs can only inhibit larvae or eggs, but have no inhibitory effect on adults.The selectivity of traditional medicine is poor, and the side effects are also large.With the use of drugs, the sensitivity of traditional drugs such as artemisinin and praziquantel decreases, resulting in a high risk of drug resistance.Target-based drug discovery is one of the most effective strategies in the field of drug development.Advancements in technological knowledge in the area of drug designing resulted in the identification of potential drug molecules.A reasonable drug design method is to find and design a reasonable drug molecule according to the structural characteristics of drug action targets (proteins, enzymes,etc.) and compounds revealed by the basic research of drugs, so as to find a lead compound that can selectively act on the target and has pharmacological activity.
Schistosomiasis is a parasitic disease that affects hundreds of millions of individuals worldwide.Praziquantel is the only drug recommended by the WHO to treat human schistosomiasis.Therefore, it is crucial to develop drugs to treat this disease.SmHDAC8and TGR have become two potential targets for the design and development of schistosome inhibitors.The aromatic hydroxamic acid compounds J1075 and TH65 have become the leading compounds of excellentsmHDAC8 inhibitors.Because the inverted conformation of F151 in the structure ofsmHDAC8 is highly specific for schistosomase, researchers can turn their research sites to H292 residues.At the same time, studies have shown that the oxadiazole-2-oxides can effectively inhibit TGR and improve the pharmacokinetics and bioavailability of TGR inhibitors, which is a desirable research direction in the future.
HAT, Chagas disease and leishmaniasis are caused by three trypanosoma species (Trypanosoma brucei,Trypanosoma cruziandTrypanosomaleishmaniasis), which are among the most deadly NTDs.The 4-aminoquinoline group has been proved to have insecticidal and antimalarial activities, so the introduction of 4-aminoquinoline group into the design of novel trypanosoma inhibitors has become a desirable research direction.Metabolic pathways of potential drug targets have opened up a new research field, and influencing glycolysis, gluconeogenesis and pentose phosphate pathway in trypanosoma is a strategy that can be used to develop effective antitrypanosomiasis strategies.
Filariasis is a common vector-borne tropical disease.Currently,the commonly used drugs in clinical practice are not effective in killing imago of filarial, and there are still no novel drugs for clinical use.Calumenin and DHFR were proved to be specific drug targets for filariasis.The virtual screening results showed that ITC was an ideal calmodulin specific compound of nematodes.In addition, the mechanism of DHFR inhibitors may treat filariasis through apoptosis, which can guide research backbones to further study filariasis drugs.Toxoplasmosis is a common zoonotic parasitic disease.However, it is very important to explore new drugs for the treatment of toxoplasmosis because of the high price and side effects.Sulfone bisphosphonate derivative have greatly improved the inhibitory effect onT.gondiicompared with the reported compounds, which can be further optimized to obtain more effective antitoxoplasma drugs.
In this paper, we reviewed the new targets such assmHDAC8,fumarase target, TGR,TcGlcK and PFK involved in the main tropical disease pathogens such as schistosomiasis, HAT, Chagas disease, leishmaniasis, filariasis and toxoplasmosis in recent years.At the same time, the pharmacological effects of typical lead compounds corresponding to each disease, the structural characteristics of the mother nucleus and the pharmacological activities of the substituents were analyzed in detail.Although the activity of most new compounds has been higher than that of standard drugs,they have not been approved for clinical use.From our point of view, optimizing existing structures, finding new targets or combination therapy can solve the current situation that leads to the lack of preclinical and clinical results.When studying the active sites of different targets, we deeply explored the interaction mechanism between the lead compound and the target, and found that the multi-target design method can open up a new way for finding new small molecular compounds of antiparasitic diseases.In this work, we compiled the currently reported inhibitors against parasitic diseases as far as possible, in order to provide new research ideas for potential antiparasitic compounds.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work is funded by the National Natural Science Foundation of China (No.82073311), the National Key Research and Development Program of China (No.2020YFC2005500), the Natural Science Foundation of Sichuan Province (No.2022JDTD0025), Science and Technology Project in Chengdu of Sichuan Province of China (No.2022-YF05–01620-SN), the Key Research and Development Program of Science and Technology Department of Sichuan Province(No.2019YFS0514), the Clinical Research and Transformation Fund of Sichuan Provincial People’s Hospital (No.2021LZ03), Open Fund of the State Key Laboratory of Traditional Chinese Medicine Resources in Southwest China (No.2021HX026), the State Administration of Traditional Chinese Medicine (No.JDZX2015210), the Open Research Fund of Chengdu University of Traditional Chinese Medicine Key Laboratory of Systematic Research of Distinctive Chinese Medicine Resources in Southwest China (No.2018GZ2011005),Screening and evaluation of anti-hepatic fibrosis varieties in traditional Chinese medicine formula granules (No.2022HX006), and the Special Fund of Sichuan Provincial Administration of Traditional Chinese Medicine (No.2021MS276).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2023.108359.
 Chinese Chemical Letters2023年10期
Chinese Chemical Letters2023年10期
- Chinese Chemical Letters的其它文章
- Tribute text in memoriam of James N.Seiber (1940–2023)
- Recent advances in MXenes-based glucose biosensors
- Oxidative cyclopalladation triggers the hydroalkylation of alkynes✩
- An integrated supramolecular fungicide nanoplatform based on pH-sensitive metal–organic frameworks
- Probing the effect of nitrate anion in CAN: An additional opportunity to reduce the catalyst loading for aerobic oxidations✩
- Nickel-catalyzed reductive coupling reaction of monofluoroalkyl triflates with alkyl carboxylic acids toward the synthesis of α-alkyl-α-fluoro-alkylketones✩