
Computational investigation on the molecular structure and chemical reactivity of a traditional Chinese medicine extract MK-1 molecule
2023-11-16 07:04:48JianZhang
Jian Zhang
School of Chemistry and Material Science,Shanxi Normal University,Taiyuan 030032,China
Abstract MK-1 molecule (C16H16O2),the simplest structure of vitamin K (VK) compound family,is an extract from traditional Chinese medicine Cymbopogon distans (Nees ex Steud.) Wats (Chinese name YunXiangCao),which has attracted a great deal of attention in recent years due to its antiasthmatic,antitussives and expectorant effects.To investigate the molecular structure and chemical reactivity of MK-1 molecule,computational investigations on six conformational minima structures were carried out at the MP2/6-311++G(2d,2p) level of theory.Several local reactivity descriptors including condensed Fukui function,average local ionization energy,and molecular electrostatic potential on each individual atom were determined to predict the intrinsic reactivity of MK-1 molecule.
Keywords: traditional Chinese medicine extract;MK-1 molecule;molecular structure;chemical reactivity
1 Introduction
Throughout history,traditional Chinese medicine has used many natural grasses to safeguard public health.Among them,Cymbopogon distans(Nees ex Steud.) Wats (Chinese name Yunxiangcao) is a perennial aromatic grass growing wild in China,which has attracted a great deal of attention due to its antiasthmatic,antitussives and expectorant effects.MK-1 molecule (C16H16O2),the simplest structure of vitamin K (VK)compound family,is the main active constituent ofCymbopogon distans(Nees ex Steud.) Wats.VK has two naturally occurring forms,VK1(phylloquinone)manufactured by plants and mainly found in green leafy vegetables and VK2(menaquinone) produced by bacteria and found in fermented products and in foods of animal origin.Besides,there is a synthetic form VK3(menadione),which is generally used as a VK source in commercial feeds for domestic animals [1-4].All of these forms share the 2-methyl-1,4-naphthoquinone ring structure,but the carbon chain substituted at the 3-position is different.VK is classified into MK-1 molecule -MK-14 molecule according to the length of the carbon chain,where the number stands for the unit of prenyl substituent.MK-1 molecule is regarded as the model molecule representing VK compound family,because VK1has exactly the same ring structure and the first unit of the substituent chain as VK2.Therefore,the theoretical investigation on the MK-1 molecule actually represents the investigation on both VK1and VK2forms.
MK-1 is a cofactor in posttranslational carboxylation of proteinbound glutamate residues,which are converted intoγ-carboxyl glutamic acid(Gla) [5].The 1,4-naphthoquinone ring structure of MK-1 contributes to the antioxidative properties [6].Strong charge-transfer interactions of MK-1 with thiazine derivatives,antimalarials quinine and quinidine are observed [7].MK-1 goes through chemically reversible electron transfer (ET)processes that are often coupled with proton transfer(PT) reactions.The reduction of MK-1 occurs in a two proton/two electron (2H+/2e-) transfer which is commonly known as proton coupled electron transfer(PCET) reaction to form the MK-1 hydroquinone.Subsequently,it is oxidized back to MK-1 via a MK-1 epoxide intermediate [8,9].A lot of experimental studies on MK-1 were conduced owing to its significance of biological importance [10-16].
In addition to the optimization of experimental methods,the development of computer hardware,the development of quantum chemistry,new computational models and algorithms,and userfriendly interfaces have reduced the obstacles of computation in the discovery and structure elucidation of natural products.Consequently,computational chemistry software has become a common tool to discover and determine the structure of natural products in recent years.There is a trend to use a smaller number of comprehensive commercial software packages,such as GAUSSIAN,to carry out a larger proportion of computational chemistry research.Gaussian software provides new methods for studying molecular systems in chemistry.GaussView software provides a rich set of construction and visualization functions.
Unfortunately,up to now,theoretical investigation on the chemical reactivity is scarce,resulting in the lack of information on the intrinsic reactivity of MK-1.In order to present a full description of the active sites of MK-1,this study reports the local reactivity descriptors calculations in the computational work,which are not available in the exiting literature but are necessary for understanding the chemical reactivity of MK-1.
Six conformational minima structures were observed in potential energy surface study by dihedral angle scanning [17,18].The geometries of six minimum energy structures of MK-1 were fully optimized at the second-order Møller-Plesset(MP2) perturbation level of theory [19] with the 6-311++G(2d,2p) basis set using the Gaussian 09 Revision E.01 [20].Frequencies of the optimized structures of these conformers were calculated,and the absence of any imaginary value confirmed that these structures were true energy minima.It is necessary to use the DENSITY=MP2 option in the calculation to restore the proper spin density.The condensed Fukui function was plotted with the aid of GaussView Revision 6.0.16 [21].The average local ionization energy and molecular electrostatic potential were calculated using the Multiwfn Revision 3.8(dev) [22] and molecular graphics program VMD Revision 1.9.4a53 [23].
2 Materials and methods
Several important local reactivity descriptors including condensed Fukui function,average local ionization energy and molecular electrostatic potential have been proposed over the last 3 decades,which characterize the chemical reactivity and site selectivity in chemical reactions.Local reactivity descriptor is an important tool for the chemists,which is suitable for the prediction of non-periodic systems.
The Fukui function (FF) introduced by Parr and Yang [24] is based on the spirit of Fukui’s frontier molecular orbital theory (FMO) and defined as the following first-order partial derivative:
where,FF can be interpreted as the change of the electron densityρ(r) when the total number of electrons of the systemNis changed.It can also be interpreted as the change of the sensitivity of chemical potentialμwhen the external potentialV(r) is changed.On this basis,discontinuities inρ(r)versusNplot provide 3 different types of FF at a frozen molecular geometry:
where,right-hand-side derivativef+(r)corresponds to nucleophilic attack,indicating that one electron in the system increases (N→N+1).Meanwhile left-hand-side derivativef-(r)corresponds to electrophilic attack,indicating that one electron in the system decreases (N→N-1).f0(r) is obtained by the arithmetic average of the previous two derivatives.Compared wtih other regions in the molecule,the region with a high FF value is a favorable reactive site.A simple method to analyze Fukui function called condensed Fukui function is used as a descriptor in quantitative structure-activity relationships and calculated using atomic population analysis method and defined as following 3 types:
wher,eqAis the charge of an atom A.Nis the number of electrons in the neutral molecule,whileN+1 andN-1 correspond to anion and cation respectively.
The average local ionization energy (ALIE)introduced by Sjoberg et al.and Politzer et al.is another tool to determine the chemical reactivity of the electronic charge in a molecule.It can be defined by equation [25,26]:
where,ρi(r) is the electron density of atomic or molecular orbitali,its orbital energy isεi,andρ(r) is the molecular total electron density.The summation is over all occupied atomic or molecular orbitals.The ALIE can be explained as the average requiring energy to remove an electron at any point in the atomic or molecular space.The region with the lowest ALIE value on molecular surface is the most favorable site for interactions with electrophiles.
Electrostatic potential (ESP) is a widely employed visualization method to understand the static charge distribution for analyzing and predicting the sites of chemical reactivity within the molecule.With position vectorr,it is defined by a molecule possessingNnuclei in the following way [27]:
where,ZAis the charge on nucleus A whose position isRA,ρ(r') is the corresponding continuous electron density of the molecule,andr'is a dummy integration variable.
3 Results and discussion
3.1 Geometrical structure and thermodynamic energy
The six local minima conformations of MK-1 molecule were optimized at MP2/6-311++G(2d,2p) level of theory and illustrated in Fig.1.The characteristic torsion angles,gas-phase relative thermodynamic energies and the HOMOLUMO energy separations for MK-1 conformers are reported in Table 1.The isomerization depends on the orientation of the bond C13-C14 around the bond C12-C13,as well as the orientation of the group C12-C13-C14 around the bond C10-C12.The internal rotation of bond C10-C12 yielded three minima,and the torsional angles C7-C10-C12-C13 are approximately 88°,91° and 122°.For bond C12-C13,there is one distinct minimum with the torsional angle C10-C12-C13-C14 of 122° when C7-C10-C12-C13=88°,along with C10-C12-C13-C14 of 110° when C7-C10-C12-C13=91° and C10-C12-C13-C14 of 63° when C7-C10-C12-C13=122°.The values obtained at MP2/6-311++G(2d,2p) level indicate that there is no significant energy difference between these conformers and the differences in thermodynamic energies for these conformers are less than 1 kcal/mol.The conformers 2 and 4 are the most stable form,since the approximation MP2/6-311++G(2d,2p) yielded the sequence of relative thermodynamic energies: 2=4 <1=5 <3=6.The most stable conformation for all these conformers is the molecular structure with the consecutive torsional angle of 110° and 90°.The HOMO-LUMO gaps were calculated by the energy difference between the HOMO and LUMO Kohn-Sham (KS)orbitals [28,29].A larger HOMO-LUMO gap implies higher kinetic stability as it is energetically disadvantageous to excite electron into LUMO or to extract electron from HOMO.Therefore,the chemical reactivity for these conformers is:1=5 <2=4 <3=6.

Table 1 The characteristic torsion angles (º),gas-phase relative thermodynamic energies (kcal/mol) and HOMOLUMO energy separations (eV) for six conformational minima structures of MK-1

Fig.1 Six conformational minima structures of MK-1
3.2 Condensed Fukui function
The sites with higher condensed Fukui function values are more reactive centers in molecular systems.In order to reveal detailed information about the reactive sites,the condensed FF analysis was performed and plotted in Fig.2.The light grey color corresponds to the Fukuifunction maxima,where an attack by an electrophile,such as a proton,is expected to occur.The corresponding condensed FF values of the most stable MK-1 conformers 2 and 6 (Table 2) suggest that the O2 site is the most susceptible to electrophillic attack for conformer 2,while the C3 site is the most susceptible to electrophillic attack for conformer 6.
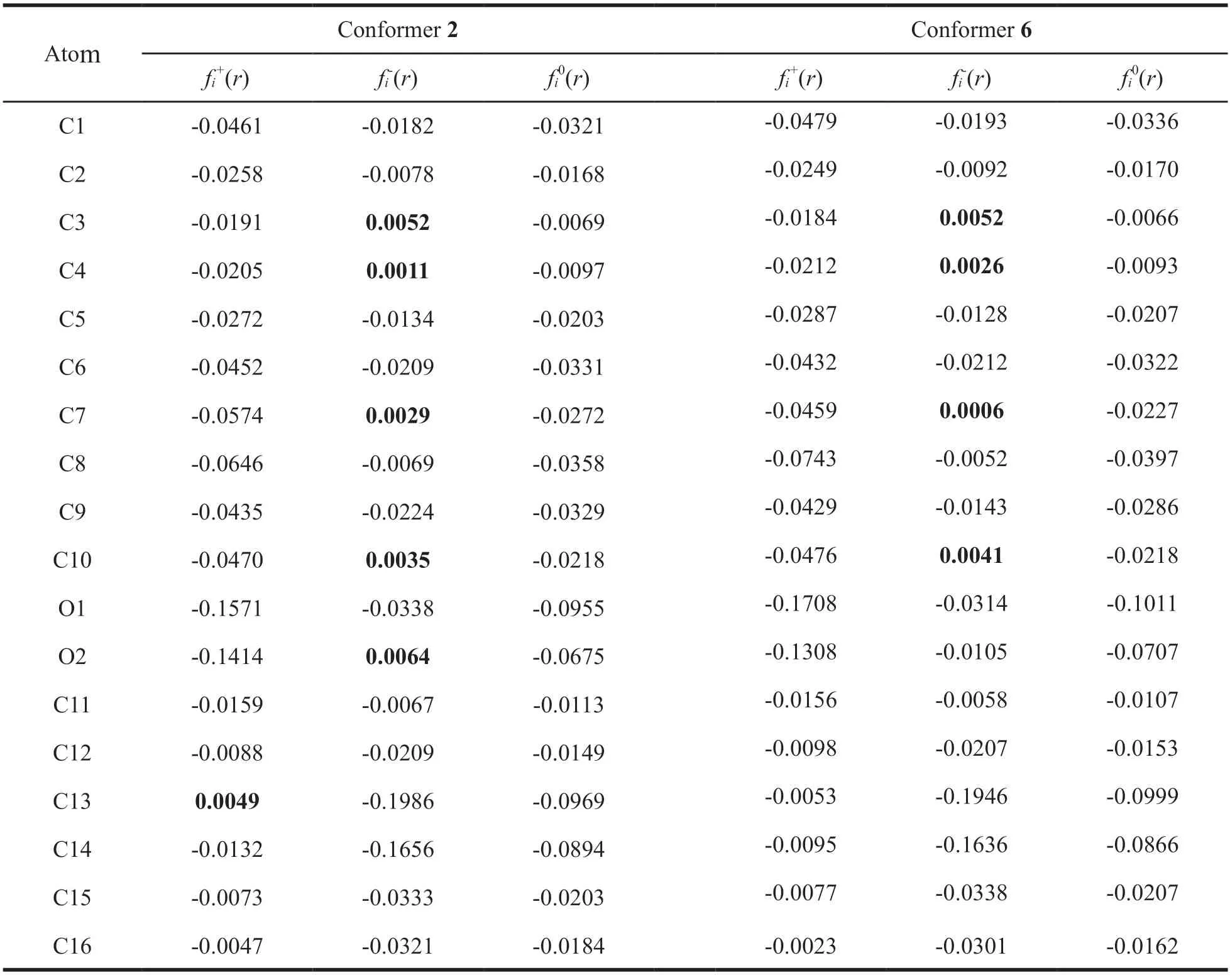
Table 2 Condensed Fukui functions for conformers 2 and 6 of MK-1
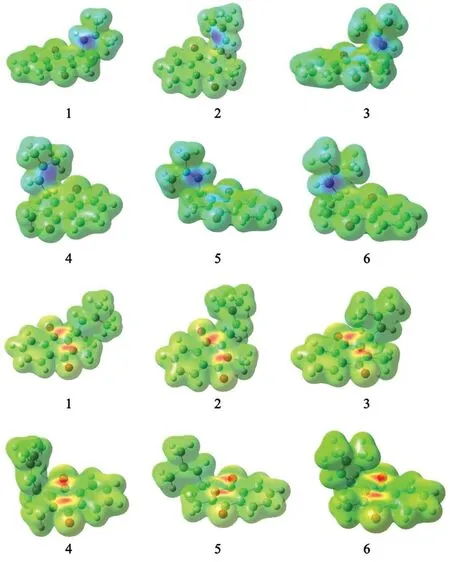
Fig.2 Condensed Fukui functions map (up: Fukui fi+(r),down: Fukui fi-(r) function) for six conformational minima structures of MK-1
3.3 Average local ionization energy
Average local ionization energy on molecular van der Waals (vdW) surface is a powerful method to analyze molecular recognition mode and to predict the reactive sites and surface reactivity.It is extensively used to reproduce atomic shell structure and quantify local polarizability measuring electronegativity.ALIE is a more suitable descriptor than molecular electrostatic potential when it comes to the determination of molecule sites prone to electrophilic attacks.The lower the ALIE value,the weaker the binding at the point,which is predicted to be reactive site.As shown in Fig.3,the black color,which indicates the lowest values of energy needed to remove the electron,is located in the near vicinity of prenyl chain,indicating that this unit is prone to the electrophilic attacks.Black color is also located on atoms of aromatic ring on the same side of prenyl chain,indicating that this position is also prone to the electrophilic attacks,but the degree is lower than that of prenyl chain.On the other side,the highest ALIE value (red color) for the MK-1 conformers is-459 kcal/mol and it is mostly located on the other side of aromatic ring.

Fig.3 Average local ionization energy map for six conformational minima structures of MK-1
3.4 Molecular electrostatic potential
Molecular electrostatic potential has been widely used to identify the sites of nucleophilic attack and electrophilic attack.ESP mapped along with vdW surface extrema of six conformational minima structures of MK-1 is shown in Fig.4 with different colour ranges.The ESP surface can be negative or positive,with a higher negative value corresponding to the light grey color and a higher positive value corresponding to the black color.As shown in Fig.4,the largest negative values for conformer 6 were found at O1 (-28.4 kcal/mol)and O2 (-29.02 kcal/mol),which correspond to the global minimum on vdW surface.The global maximum appears in the positively charged H3 and H4,and the ESP values at these points are much larger (20.63 and 20.66 kcal/mol) than those at other points.The higher negative ESP value corresponds to a better attraction of the protons by the concentrated electron density in the molecule.Similarly,the higher positive ESP value corresponds to the better repulsion of the proton by the atomic nuclei in the region where low electron density exists and the nuclear charge is incompletely shielded.

Fig.4 Molecular ESP map for six conformational minima structures of MK-1
4 Conclusion
MK-1 molecule (C16H16O2) is an extract from traditional Chinese medicineCymbopogon distans(Nees ex Steud.) Wats (Chinese name YunXiangCao).In this study,a series of theoretical calculations have been performed on several local reactivity descriptors to predict the chemical reactivity of six conformational minima structures of MK-1.The structures of these compound were optimized using MP2 level of theory with the 6-311++G(2d,2p) basis set.The sequence of relative thermodynamic energies is conformer 2=conformer 4
