
改性微晶纤维素增质PDMS-BG生物复合材料的制备与性能研究*
2023-11-14 03:03白小转李蓓蓓
化学工程师 2023年10期
陈 茎,白小转,薛 敏,石 奇,李蓓蓓
(西安文理学院 化学工程学院,陕西 西安 710065)
近年来,人口老龄化进程逐渐加快,随之而来的骨质疏松症等骨科疾病,或者因骨肿瘤切除、交通事故引起的骨缺损日益严重,而很多无法自愈的骨缺损需要骨替代或者骨修复材料[1]。目前,骨替代和修复材料主要是金属和合金,其次是生物陶瓷、聚合物、复合材料、人体和动物骨骼及骨骼衍生物等[2-4]。但生物材料无论是在体内细胞和蛋白调控的生理环境,还是体外模拟体液的生理环境都会在材料表面发生沉积反应,生成一层类似于骨组织中的无机矿物质——碳酸羟基磷灰石。生物活性玻璃主要是由Na2O、CaO、SiO2和P2O5等基本成分组成的硅酸盐玻璃,将其植入人体骨骼缺损部位,会与骨直接结合修复骨组织[5,6]。但生物活性玻璃具有脆性大、收缩性大和强度低等缺点,因此,通过将其与高强度的可生物降解的高分子材料复合,可有效地提高材料的机械性能,如生物活性玻璃与聚乳酸、聚己内酯的复合等。但其与人体骨相比脆性大,严重限制了该类材料的使用范围,目前,主要用于骨损伤及骨料填充等非承重部位[7-9]。
微晶纤维素(microcrystalline cellulose,MCC)具有高强度、可生物降解和绿色环保等优良性能,具有较大的长径比和较完整的结晶结构,机械性能优异,将增强相——MCC 及其衍生物加入到基质中,会增加复合材料的拉伸强度、储能模量和弹性模量等性能,从而拓宽复合材料的应用范围[10,11]。但由于MCC 表面存在大量的强极性羟基基团,与疏水性的聚合物基体间的相容性很弱,会影响其在基质中的分散和增质效果,极大的限制了MCC 的应用。
本实验用硅烷偶联剂γ-(甲基丙烯酰氧)丙基三甲氧基硅烷(KH570)改性微晶纤维素表面的亲水羟基基团的方法,降低其强极性,并以生物活性的PDMS-BG 溶胶为基质,采用溶胶凝胶法制备了具有性能优异的mMCC-PDMS-BG 生物复合材料,研究改性微晶纤维素在基质中的分散性以及提高填料与基质之间的界面作用力,考察了不同含量改性微晶纤维素增质生物复合材料的影响。
1 实验部分
1.1 试剂与仪器
正硅酸乙酯(TEOS 98%)、Ca(NO3)2·4H2O(99%)、HCl(37%)、溶剂异丙醇(99.7%)、四氢呋喃(99.5%),科密欧化学试剂公司;羟基封端的聚二甲基硅氧烷(PDMS,HO[-Si(CH3)2O-]nH Mn=1100 AR 上海阿拉丁试剂有限公司);微晶纤维素(97%成都市科隆化学品有限公司);KH570(98% 国药集团化学试剂有限公司);乙醇(99.7%)、HAc(99.5%),天津市富宇精细化工有限公司。
采用德国Bruker OPUS 80V 型傅里叶红外光谱仪(FT-IR)测试样品在模拟体液(Simulated body fluid,简称SBF)中培养前后的组成和结构;采用日本(JEOL)S-4800 型场发射扫描电镜(FE-SEM)表征培养前后样品的表面形貌,并用能谱仪表征在SBF培养前后样品表面的Ca、P、Si 和C 的含量;采用日本(Rigaku)D/max-2400 型X 射线衍射仪(XRD)来测定样品相结构的图谱。
1.2 实验方法
1.2.1 PDMS-BG 的制备 将正硅酸乙酯溶于异丙醇和四氢呋喃的混合溶剂中,加入HCl 催化反应,反应20min 后加入去离子水,搅拌2h 后加入聚二甲基硅氧烷进行缩聚反应,继续搅拌获得溶液1;将Ca(NO3)2·4H2O 充分溶解于去离子水和异丙醇中,制得溶液2;将溶液1 和溶液2 混合,搅拌均匀,制得聚二甲基硅氧烷改性硅酸盐生物活性玻璃溶胶,记为PDMS-BG 溶胶。
1.2.2 改性微晶纤维素mMCC 的制备 向乙醇水溶液中加入硅烷偶联剂KH570,同时调节pH 值为3.5~4,在40℃恒温水浴搅拌1h,接着加入MCC,在40℃恒温水浴继续搅拌反应2h,然后室温静置24h,离心分离,将获得的沉淀物用去离子水洗涤,在40℃烘干后得到改性微晶纤维素,记为mMCC。
1.2.3 mMCC-PDMS-BG 生物复合材料的制备 将mMCC 加入到PDMS-BG 溶胶中,超声1h,接着搅拌15h,陈化24h 后倒入培养皿中,用保鲜膜密封,放入60℃烘箱中烘干5d,继续升温至100℃烘干24h,即可得到mMCC-PDMS-BG 生物复合材料。
1.2.4 体外生物活性培养 材料具有生物活性,是指材料植入人体后表面会形成碳酸羟基磷灰石(HCA),与骨组织发生化学键合。实验室采用和人体血浆中的离子浓度相似的模拟体液(Simulated body fluid,简称SBF)来研究材料的生物活性。SBF 模拟体液是由各类无机盐溶于水配制而成,通常用于体外模拟实验[12]。
2 结果与讨论
2.1 改性微晶纤维素结构表征
图1(a)为未改性的微晶纤维素SEM 图谱,如图所示为团簇状,聚集比较严重。图1(b)为改性的微晶纤维素SEM 图谱,经过硅烷偶联剂KH570 改性后显示出分散的细微短棒状。图1(c)所示的FTIR 中,未改性的微晶纤维素MCC 呈现典型的纤维素红外特征谱图,而经过改性的微晶纤维素mMCC的红外谱图所对应的吸收峰的位置和强度和未改性微晶纤维素几乎一致,只是在1720cm-1处出现了一个新的特征峰,该峰是接枝KH570 酯基中C=O 的伸缩振动吸收峰,说明微晶纤维素被成功改性[13]。图1(d)为微晶纤维素的XRD 图谱,由XRD 图可见,改性前后的微晶纤维素特征峰没有明显的改变,这是因为硅烷偶联剂KH570 只是对微晶纤维素表面进行了改性,并未破坏微晶纤维素的晶体结构,依然保持了其短棒状结构和优良的力学性能。
2.2 mMCC-PDMS-BG 复合材料表面形貌和结构表征
图2 为制备的mMCC-PDMS-BG 溶胶和生物复合材料块体实物图。图2(a)右瓶中为未改性的微晶纤维素添加到生物活性的PDMS-BG 溶液中,可以看到MCC 分散效果欠佳,容易产生沉淀,可能是由于表面羟基发生团聚引起的,而左瓶中改性mMCC 在PDMS-BG 溶液中能稳定分散,说明改性后减弱了微晶纤维素表面的极性,从而增加了mMCC 在PDMS-BG 基体中的分散性和相容性。图2(b)是制备的mMCC-PDMS-BG 生物复合材料块体实物图,图中显示材料具有良好的弹性和韧性,说明添加适量mMCC 后使得微晶纤维素与基质界面之间的相互作用力更强。实验中发现,添加了mMCC 有助于材料形成三维网状结构,提高了材料的成型能力,并且缩短了制备周期。但随着mMCC 含量的增加会产生团聚现象,导致材料破裂现象发生(图2(c)),影响复合材料的成型性能。
采用扫面电子显微镜观察所制备的mMCCPDMS-BG 生物复合材料的表面形貌特征,结果见图3。
由图3 可见,加入不同含量改性微晶纤维素mMCC(mMCC 占总质量比为:(a)0.5wt%、(b)1.0wt%、(c)1.5wt%、(d)2.0wt%)样品表面形貌各异,(a)和(b)表面都具有均匀的孔状结构,但当mMCC 含量大于1.0 wt%时,孔结构消失,复合材料表面变得粗糙,能看到部分颗粒分布在复合材料表面(图3(c)、(d)),说明当微晶纤维素超过一定量之后会发生团聚现象。
图4 为含量1.0wt%mMCC 生物复合材料的EDS 图谱。

图4 含量1.0wt% mMCC 复合材料的EDS 图谱Fig.4 EDS patterns of composits containing 1.0wt% mMCC
由图4 可见,有少量钙和磷元素存在,说明无机相的BG 和PDMS 发生了有效的杂化复合。
图5(a)为含量1.0wt%mMCC 生物复合材料的XRD 图谱。由于基质即生物活性玻璃PDMS-BG 是典型的非晶相结构,添加了少量的改性微晶纤维素后依然表现出无定形结构。图5(b)是不同含量mMCC 的红外光谱图,800cm-1和850cm-1红外吸附带分别对应Si-C 和Si-OH 对称弯曲振动模型,1080cm-1是Si-O-Si 非对称振动伸缩模型,1260cm-1和1390cm-1分别属于Si-CH3和C-H 对称弯曲振动模型[14,15]。
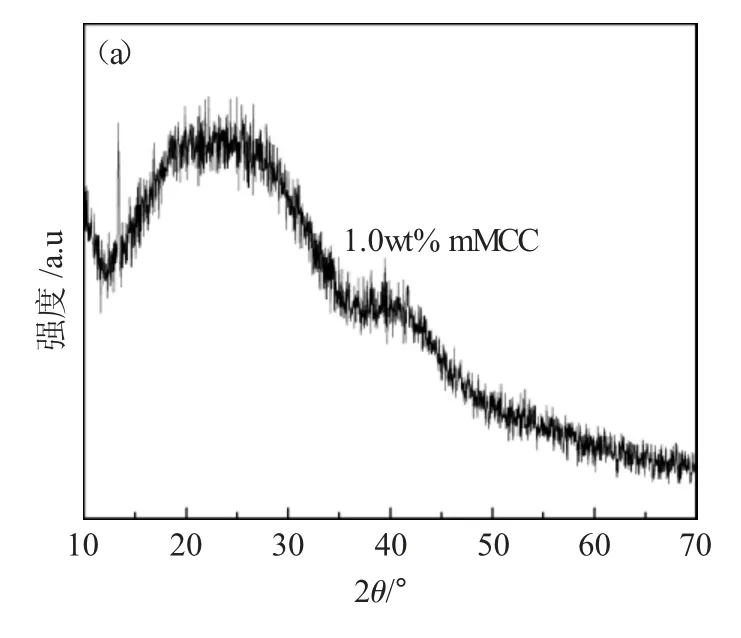

图5 (a)含量1.0 wt%mMCC 复合材料的XRD 图谱,(b)不同含量mMCC 的mMCC-PDMS-BG 生物复合材料FT-IRFig.5 (a)XRD patterns of composites with 1.0wt% mMCC,(b)FT-IR of mMCC-PDMS-BG bicomposites with different contents of mMCC
2.3 mMCC-PDMS-BG 块体材料体外生物活性的研究
不同含量的mMCC(0.5wt%、1.0wt%、1.5wt%、2wt%)对生物复合材料mMCC- PDMS-BG 的生物活性即羟基磷灰石形成能力有很大的影响。与模拟体液SBF 溶液反应3d 的复合材料的表面形貌见图6,与图3 未反应的试样相比较,所有材料的表面发生了明显的变化,均产生了一层新的沉积物,并且新生成的颗粒物覆盖了原先的孔状结构,结合后面的XRD 和FT-IR 光谱分析,这些新生产的球状颗粒可认为是羟基磷灰石颗粒。mMCC 含量大于1.0wt%时,mMCC-PDMS-BG 生物复合材料表面球状沉积物显著变少,这是因为mMCC 含量大于1.0wt%时,表面孔状结构逐渐消失,而孔状结构的存在有利于羟基磷灰石的形成。

图6 不同含量mMCC 的mMCC-PDMS-BG在SBF 中反应3d 后的SEM 图Fig.6 SEM patterns of mMCC-PDMS-BG with different contents of mMCC after soaking in SBF for 3 days
图7 为mMCC 含量为1.0wt%的mMCC-PDMSBG 生物复合材料在SBF 中反应3d 后的EDS 能谱。

图7 含量1.0wt%mMCC 的mMCC- PDMS-BG 生物复合材料在SBF 中反应3d 后的EDS 能谱Fig.7 EDS patterns of mMCC-PDMS-BG bicomposites with 1.0wt% mMCC after soaking in SBF for 3 days
由图7 可见,钙和磷的含量显著增加,说明复合材料具有良好的生物矿化能力。
为了进一步确定mMCC-PDMS-BG 生物复合材料表面的沉积物的物相组成和结构,对在SBF 溶液中反应3d 后的复合材料试样进行XRD 分析,图8(a)为含量1.0wt% mMCC 的mMCC- PDMS-BG 生物复合材料在SBF 中反应3d 后的XRD 图谱,与反应前的XRD 图谱相比,反应后的XRD 图谱发生了很大的变化。由玻璃相无定形结构变成新的衍射峰出现,在2θ 为26°、31°和39° 时,对应于羟基磷灰石衍射的200,211 和310 衍射晶面(与JCPDS 卡片进行对比),说明杂化材料均具有体外磷灰石(HA)形成能力。可进一步通过红外光谱证实材料表面形成的磷灰石结构。由图8(b)可以看出,与反应前的红外光谱相比,反应3d 后,试样在1480cm-1和1430cm-1处出现了微弱的CO32-吸收峰[15],在复合材料的表面形成了碳酸羟基磷灰石,说明复合材料具有良好的体外磷灰石形成能力,并且含量1.0wt%mMCC 的mMCC- PDMS-BG 生物复合材料具有较为显著的磷灰石形成能力。


图8 (a)含量1.0wt% mMCC 的mMCC- PDMS-BG生物复合材料在SBF 中反应3d 后的XRD 能谱,(b)复合材料在SBF 中反应3d 的FT-IR 光谱Fig.8 After soaking in SBF for 3 days(a)XRD patterns of composites with 1.0wt% mMCC,(b)FT-IR of mMCC-PDMSBG bicomposites with different contents of mMCC
由XRD、FT-IR、SEM 和EDS 等图谱可见,mMCC- PDMS-BG 生物复合材料具有较好的生物活性,即材料表面具有易矿化的能力。经过改性的微晶纤维素表面极性基团增加,有利于提升纤维和基质反应形成化学键,进一步提升微晶纤维素与基质之间的界面结合性能,从而有效改善复合材料的整体性能。
3 结论
采用溶胶凝胶法制备了mMCC-PDMS-BG 生物复合材料,通过添加不同含量改性微晶纤维素(mMCC)对凝胶成型能力和生物活性的影响进行了研究。结果发现,添加mMCC 使制备工艺变得简单,制备周期大大缩短。将mMCC- PDMS-BG 生物复合材料进行体外培养后,结合FT-IR、XRD、SEM 和EDS图谱可以确定,材料具有良好的生物活性,并且含量1.0wt%mMCC 的mMCC-PDMS-BG 生物复合材料具有较为显著的磷灰石形成能力。mMCC- PDMS-BG生物复合材料有可能成为一种新型的骨修复或骨替代材料,或者可作为一种新型药物载体材料。
猜你喜欢
建材发展导向(2021年11期)2021-07-28
纺织科技进展(2021年3期)2021-06-09
陶瓷学报(2021年1期)2021-04-13
湿法冶金(2019年5期)2019-10-18
超硬材料工程(2016年1期)2016-02-28
湖北科技学院学报(医学版)(2015年3期)2015-02-28
深圳大学学报(理工版)(2015年5期)2015-02-28
四川师范大学学报(自然科学版)(2015年1期)2015-02-28
应用化工(2014年11期)2014-08-16
食品工业科技(2014年9期)2014-03-11
