
GEO基因芯片分析结合网络药理学及分子对接技术探析“黄连-木香-肉豆蔻”组方治疗溃疡性结肠炎的分子机制
2023-11-12 05:11:16朱文瑞王铁烽徐洪锋
世界华人消化杂志 2023年20期
朱文瑞,王铁烽,徐洪锋
朱文瑞,王铁烽,徐洪锋,绍兴市中医院中药剂科 浙江省绍兴市 312000
0 引言
溃疡性结肠炎(ulcerative colitis,UC)是一种以腹痛、腹泻、脓血便等肠道症状为主要临床表现的慢性非特异性肠道炎症性疾病[1].目前,UC的西药治疗以5-氨基水杨酸、糖皮质激素、免疫抑制和生物制剂为主,但长期应用易出现药物不良反应、药物依赖及停药后易复发等问题[2,3],而类似粪菌移植[4]、干细胞移植[5]等治疗手段多仅在临床起步阶段,价格昂贵且疗效欠稳定.中医药治疗UC有其特有的优势[6],从中医药宝库中挖掘安全有效的UC治疗手段迫在眉睫.“黄连-木香-肉豆蔻”(Huanglian-Muxiang-Roudoukou,HMR)组方出自经典名方豆蔻香连丸,据记载可用于“治泄泻,腹痛,不拘寒热赤白.”,其治疗症状与UC的临床表现大体相符.进一步通过数据挖掘分析姚乃礼等[7-9]全国名老中医在治疗UC方面的用药规律,结果发现黄连、木香、肉豆蔻是临证治疗中的高频药物.姚乃礼教授[10]、谢晶日教授[11]在治疗UC的经验方中,用HMR以清肠燥湿,涩肠止泻.综上所述,HMR是中医药治疗UC的常用药物组合,但其治疗UC的主要成分及潜在作用机制尚不明确,导致其在临床中的使用受到限制.中药复方成分多,靶点复杂,既往逐一靶点验证的研究方式太过于繁琐,需要消耗大量的人力及物力,无法全面性地探索药物机制.基因表达谱、网络药理学等技术的出现,使系统全面分析探索中药治疗疾病的机制实现了可能.因此,本文利用基因表达综合数据库(gene expression omnibus,GEO)中的芯片数据挖掘与UC发病相关的特异性表达基因,获取健康人群和UC患者的核心差异表达基因,再运用网络药理学和分子对接技术预测HMR治疗UC的可能作用机制,以期为该组方的进一步研究与临床应用提供依据.
1 材料和方法
1.1 材料 UC差异表达基因分析:在GEO数据库(https://www.ncbi.nlm.nih.gov/geo/)中以“ulcerative colitis”为关键词检索与UC相关的基因表达谱数据,选取人类正常样本及UC患者样本均大于10的GEO数据芯片,下载其对应的结果矩阵(Matrix)与平台注释文件(Platforms),数据整理分组后导入R软件,利用Limma包以|lgFC|>1(FC表示差异倍数)、P<0.05为条件筛选出差异表达基因(differentially expressed genes,DEGs),并分别绘制差异基因火山图和排名前40位差异基因的热图.
1.2 方法
1.2.1 HMR中活性成分及靶点筛选:利用中药系统药理学数据库与分析平台(traditional chinese medicine systems pharmacology database and analysis platform,TCMSP)分别以“肉豆蔻”、“黄连”及“木香”为关键词搜集HMR中各个中药的化学成分,设定口服生物利用度(oral bioavailability,OB)≥30%和类药性指数(drug-likeness,DL)≥0.18为条件,筛选出符合条件的活性成分和其所对应的靶点信息.借助UniProt数据库(https://www.uniprot.org/),限定物种为“Homo sapiens”,进行基因名匹配及标准化处理.
1.2.2 “中药-活性成分-疾病-靶点”网络的建立:将筛选得到的药物靶点基因与疾病差异基因相互映射,得到交集基因,并查找与之相对应的有效成分,将搜集到的中药、活性成分、疾病、靶标蛋白中的基因依次输入,利用Cytoscape 3.9.1软件进行可视化处理,构建“药物-活性成分-靶点”网络.
1.2.3 蛋白互作网络(protein-protein interaction,PPI)的建立:将“1.2.2”项得到的交集基因导入STRING(https://string-db.org/)数据库,物种选择人类,设置相互作用阈值为“medium confidence(置信度>0.400)”,隐藏没有相互联系的节点,将蛋白互作关系结果通过Cytoscape3.9.1软件中进行可视化处理,构建PPI网络,借助cytoCNA插件进行拓扑结构分析,计算节点的度值(Degree)、介度中心性(betweenness centrality,BC)和紧密中心性(closeness centrality,CC)值,并以Degree、BC和CC的中位数值作为节点重要性的量化参考[12,13],筛选核心靶点.
1.2.4 生物学功能富集分析:将“1.2.2”项得到的交集基因数据导入Metascape数据库(http://metascape.org/),设置P<0.01,限定物种为“Homo sapiens”,进行基因本体论(gene ontology,GO)富集分析和京都基因和基因组百科全书(kyoto encyclopedia of genes and gnomes,KEGG)富集分析.
1.2.5 分子对接验证:从PDB数据库(https://www.rcsb.org/)中下载核心靶点蛋白质结构,利用PyMOL 2.2软件除去水分子,分离原配体,保存后导入Autodock Tools 1.5.6软件中加氢、计算总电荷、设置原子类型,保存为“pdbqt”格式.从TCMSP数据库中下载上述筛选得到的核心成分(配体)的mol2结构,利用Autodock Tools 1.5.6软件设置可旋转键后保存为“pdbqt”格式文件,最后利用Autodock-vina 1.1.2软件进行分子对接,在PyMOL下实现对接结果可视化,绘制对接相互作用模式图.
统计学处理采用R4.1.2软件进行数据统计分析,采用单因素方差分析组间差异,组间两两比较用最小显著性差异法,P<0.05为差异有统计学意义.
2 结果
2.1 UC差异基因分析 从GEO数据库中选择编号为GSE87473基因表达谱数据.该芯片数据所采用的平台为GPL13158[HT_HG-U133_Plus_PM] Affymetrix HT HGU133+PM Array Plate,包含了21例健康受试者和87例成人UC患者的结肠黏膜组织样本,以|lgFC|>1、P<0.05为条件共筛选出差异表达基因967个,其中上调基因619个下调基因348个.筛选所得差异基因即为UC相关的基因差异基因,UC差异基因的火山图及热图见图1.
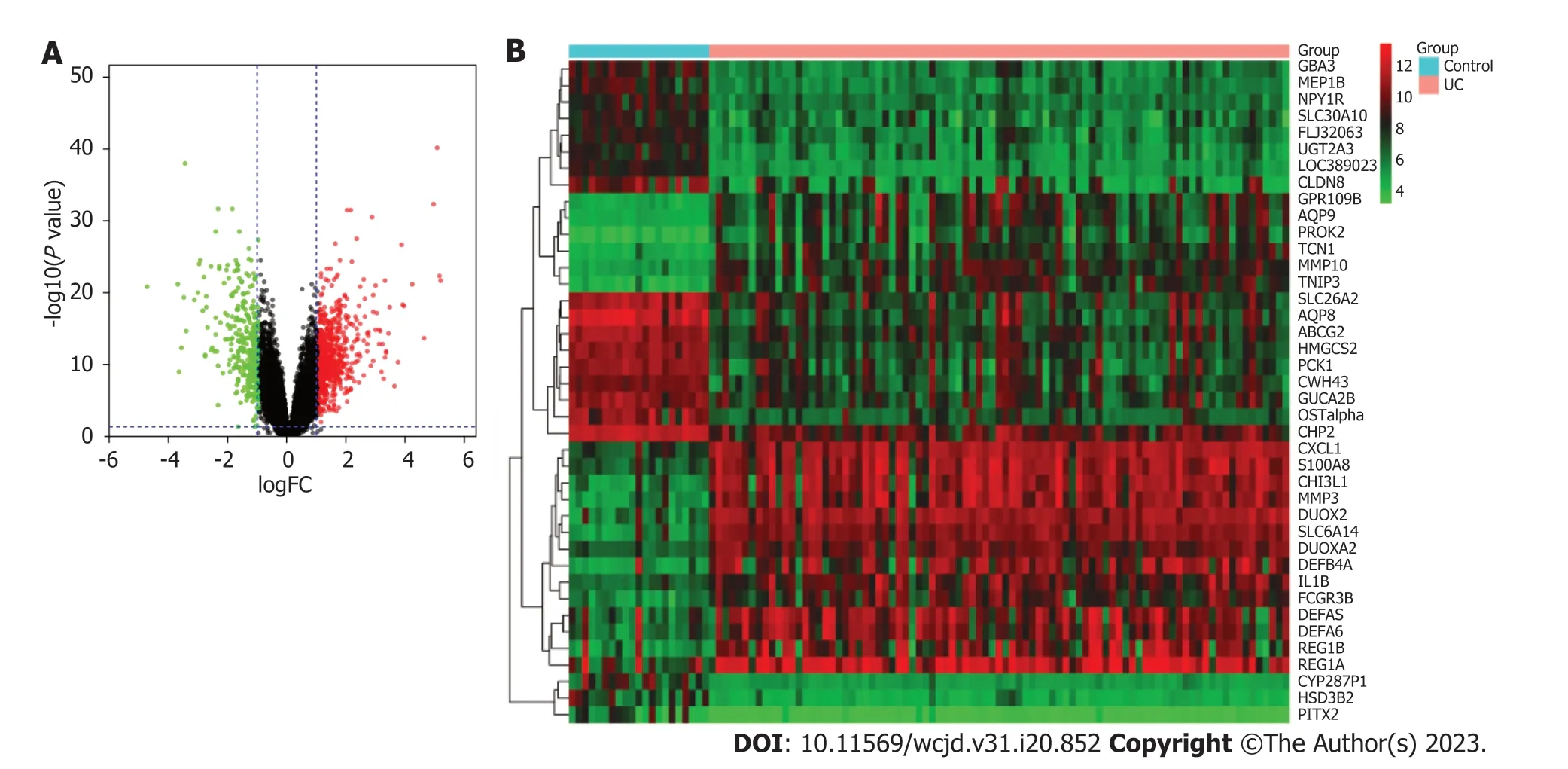
图1 UC差异基因分析.A:火山图;B:热图.红色:上调基因;绿色:下调基因;黑色:无差异基因;UC:溃疡性结肠炎;FC:差值倍数.
2.2 活性成分筛选及其作用靶点预测 根据限定条件,从TCMSP数据库收集到肉豆蔻主要活性成分9个、木香主要活性成分6个、黄连主要活性成分14个,共得到HMR的主要活性成分29个,见表1.根据筛选所得的活性成分,获得其所对应的靶点输入UniProt数据库,删除重复靶点后共得到163个有效靶点基因.

表1 HMR主要活性成分信息
2.3 “中药-活性成分-疾病-靶点”网络的建立 将药物靶点基因与疾病差异基因相互映射,共得到28个交集基因,并查找与之对应的有效成分,共得到24个有效成分,其中黄连10个、木香6个、肉豆蔻8个.运用Cytoscape软件构建“中药-活性成分-疾病-靶点”网络,见图2.图中共有54个节点和111条边线,运用网络拓扑结构分析得到各节点Degree值,其值越大意味着该节点在网络中发挥越重要的作用.通过计算结果可知:Degree值排名前5位的活性成分为槲皮素(quercetin)、豆甾醇(stigmasterol)、berberine(小檗碱)、beta-sitosterol(β-谷甾醇)、palmatine(巴马汀).

图2 HMR治疗UC的“中药-活性成分-疾病-靶点”网络图.HMR:黄连-木香-肉豆蔻;UC:溃疡性结肠炎;MMP9:基质金属蛋白酶9;MMP3:基质金属蛋白酶3;MMP1:基质金属蛋白酶1;GJA1:缝隙连接蛋白α;IL-6:白介素6;IL-1β:白介素1β;ICAM-1:细胞间黏附分子-1;Fos:原癌基因蛋白;EGF:表皮生长因子;DUOX2:双氧化酶;CXCL2:CXC趋化因子配体2;CXCL11:CXC趋化因子配体11;CXCL10:CXC趋化因子配体10;CCL2:CC趋化因子配体2;BCL2:结合蛋白B细胞淋巴瘤/白血病-2;VCAM1:血管细胞粘附分子1;TNF:肿瘤坏死因子;THBD:血栓调节素;STAT1:信号传导转录激活因子1;SPP1:分泌磷蛋白1;SELE:E选择素;PTGS2:环加氧酶2;PPARg:过氧化物酶体增殖物激活受体γ;PLAU:尿激酶型纤维蛋白溶酶原激活因子;PCOLCE:前胶原C-蛋白酶增强蛋白;NR3C2:核受体亚家族C组成员2;NOS2:诱导型一氧化氮合酶;NFKBIA:核因子κB抑制剂α.
2.4 PPI网络的建立 基于STRING数据库(https://www.string-db.org/)分析HMR治疗UC的靶点蛋白之间的相互作用,得到的PPI网络,见图3.通过Cytoscape软件及内置的Cyto NCA模块进行可视化呈现及网络拓扑分析.图中节点表示靶点蛋白,边表示蛋白之间的关联,Degree值越大,则对应的节点越大和颜色越浓.该PPI网络图中共涉及26个节点、224条边,经拓扑学分析结果后,以Degree、BC和CC的均大于其相应的中位数值为条件[12-14],并按照Degree值由大到小排序,筛选出白介素1β(interleukin-1β,IL-1β)、白介素6(interleukin-6,IL-6)、CC趋化因子配体2[chemokine (C-C motif) ligand 2,CCL2]、肿瘤坏死因子(tumor necrosis factor,TNF)及基质金属蛋白酶9(matrix metallopeptidase 9,MMP9)共11个核心靶点.
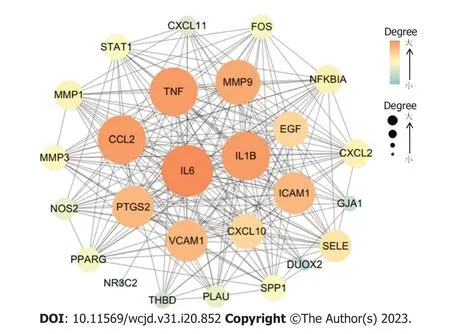
图3 HMR治疗UC的PPI网络.HMR:黄连-木香-肉豆蔻;UC:溃疡性结肠炎;PPI:蛋白互作;Degree:度值;MMP9:基质金属蛋白酶9;MMP3:基质金属蛋白酶3;MMP1:基质金属蛋白酶1;GJA1:缝隙连接蛋白α;IL-6:白介素6;IL-1β:白介素1β;ICAM-1:细胞间黏附分子-1;Fos:原癌基因蛋白;EGF:表皮生长因子;DUOX2:双氧化酶;CXCL2:CXC趋化因子配体2;CXCL11:CXC趋化因子配体11;CXCL10:CXC趋化因子配体10;CCL2:CC趋化因子配体2;VCAM1:血管细胞粘附分子1;TNF:肿瘤坏死因子;THBD:血栓调节素;STAT1:信号传导转录激活因子1;SPP1:分泌磷蛋白1;SELE:E选择素;PTGS2:环加氧酶2;PPARg:过氧化物酶体增殖物激活受体γ;PLAU:尿激酶型纤维蛋白溶酶原激活因子;NR3C2:核受体亚家族C组成员2;NOS2:诱导型一氧化氮合酶;NFKBIA:核因子κB抑制剂α.
2.5 生物学功能富集分析 运用Metascape数据库将28个药物靶点基因与UC疾病差异基因的交集基因进行GO功能富集分析,主要包括生物过程(biological process,BP)、分子功能(molecular function,MF)、细胞组分(cellular component,CC),筛选各项排列前5的条目所对应靶点具有的功能信息,绘制气泡图,见图4.由图4可知,HMR治疗UC的BP主要涉及脂多糖刺激变化过程、对神经炎症反应调节、对辐射的反应、对肽的反应、对细胞外刺激的反应等;MF主要涉及细胞因子活性、整合素结合、丝氨酸型内肽酶活性、DNA结合转录因子结合、血红素结合等;CC主要涉及质膜外侧面、膜筏、细胞外基质、RNA聚合酶Ⅱ转录调控复合物、内质网管腔等.运用Metascape数据库进行KEGG功能富集分析,筛选排列前10的条目所对应靶点具有的功能信息,绘制气泡图,见图5.由图5可知,HMR治疗UC靶点关联的通路主要有TNF信号通路、核转录因子κB(nuclear factor kappa-B,NF-κB)通路、Toll样受体(toll-like receptor,TLR)信号通路等.
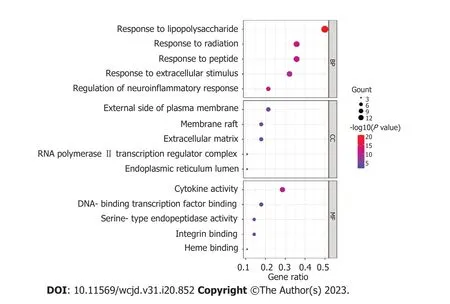
图4 HMR治疗UC的GO分析.HMR:黄连-木香-肉豆蔻;UC:溃疡性结肠炎;GO:基因本体;BP:生物过程;MF:分子功能;CC:细胞组分.
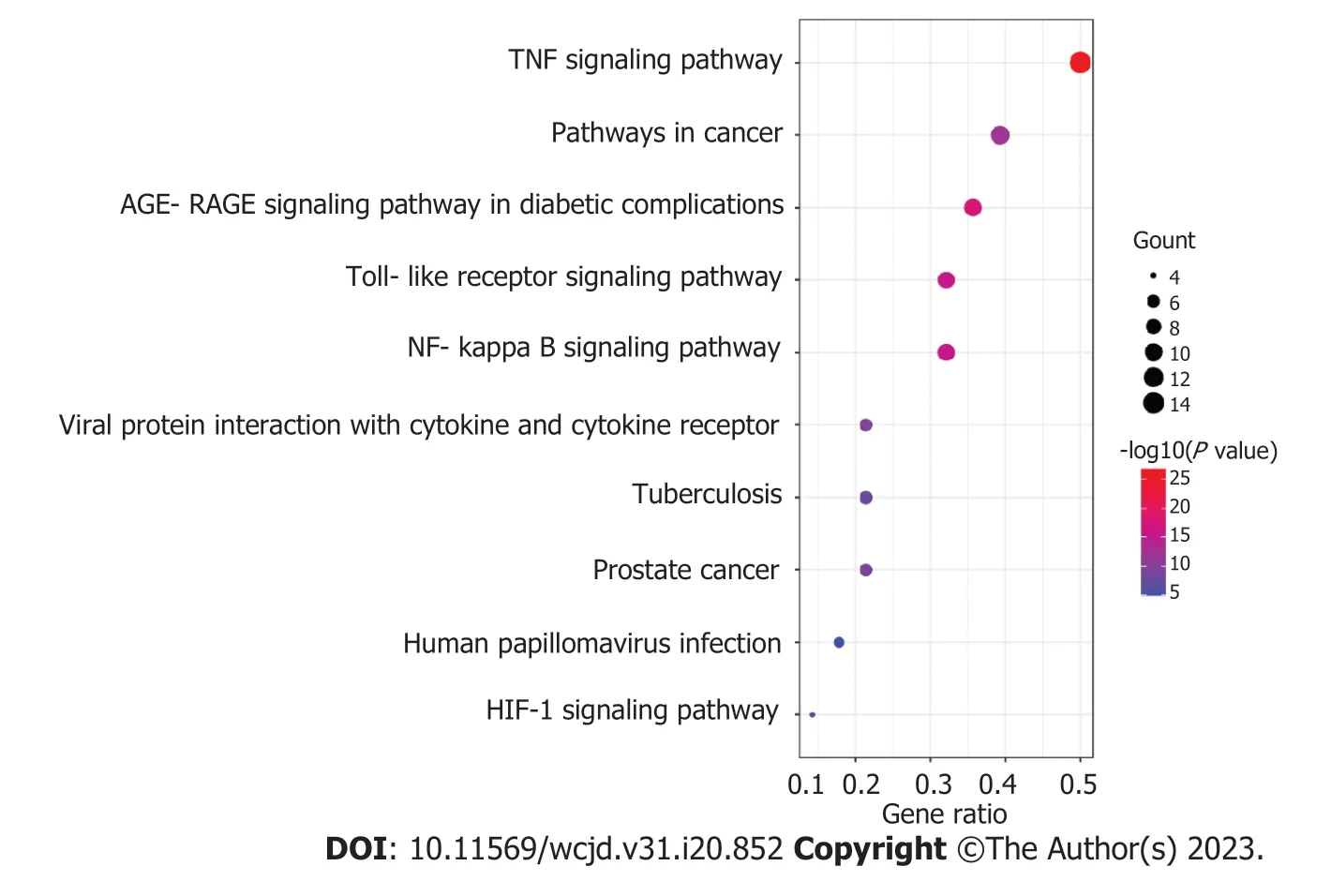
图5 HMR治疗UC的KEGG分析.HMR:黄连-木香-肉豆蔻;UC:溃疡性结肠炎;KEGG:京都基因与基因组百科全书;TNF:肿瘤坏死因子;HIF-1:缺氧诱导因子1.
2.6 分子对接验证 将“2.4”项下“中药-活性成分-疾病-靶点”网络中Degree值排名前5的活性成分,与“2.5”项下PPI网络中Degree值排名前5的核心靶点进行分子对接,计算活性成分与核心靶点间的结合能,结果见表2.结合能值越小表示结合能越高,活性成分越容易与蛋白结合,结合能-5.0 kcal/mol表明两者之间结合活性较好;小于-7.0 kcal/mol表明两者之间有很强结合活性[15].然后再分别选取结合能最低的成分,并用PyMol软件对其对接结果进行可视化展示,见图6.
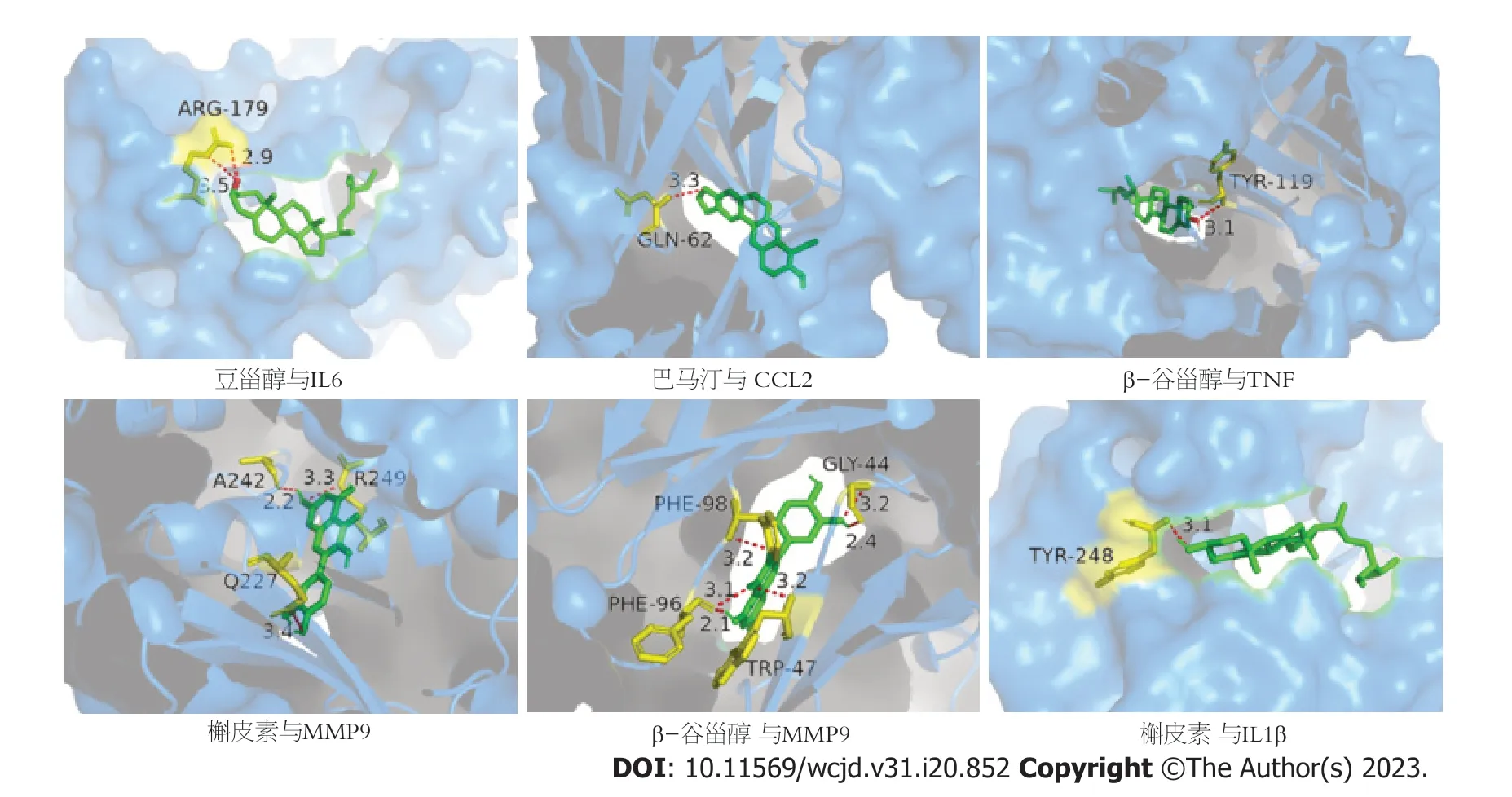
图6 活性成分与关键靶点的分子对接模式.绿色:配体;黄色:与配体对接的氨基酸残基;红色虚线:氢键;IL-1β:白介素1β;IL-6:白介素6;CCL2:趋化因子配体2;TNF:肿瘤坏死因子;MMP9:基质金属蛋白酶9.
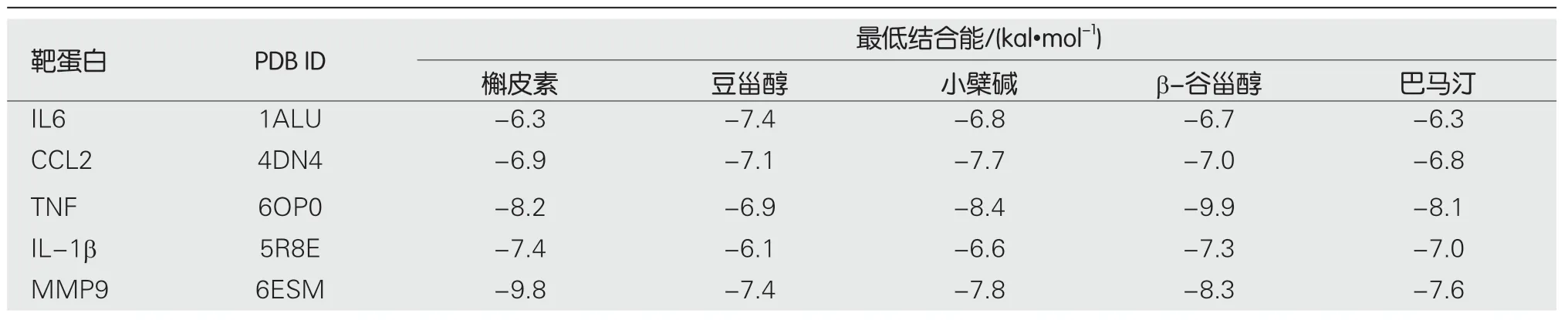
表2 分子对接结果
对接结果显示,槲皮素、豆甾醇、小檗碱、β-谷甾醇、巴马汀与关键靶点IL6、CCL2、TNF、IL-1β、MMP9的结合能均小于-5.0 kcal·mol-1,说明活性成分与靶点之间具有较好结合活性,槲皮素与MMP9,β-谷甾醇与TNF的结合能小于9.0 kcal·mol-1,说明两者之间具有强烈的结合活性.分子对接可视化分析研究发现,槲皮素通过氨基酸残基Q227、A242、R249与MMP9形成3条氢键,β-谷甾醇通过氨基酸残基TYR-119与TNF形成1条氢键,β-谷甾醇通过氨基酸残基GLY-44、PHE-96、PHE-98、TRP-47与MMP9形成4条氢键,并且上述配体化合物均能良好地包埋在受体靶蛋白的活性口袋中.
3 讨论
中药复方具有多成分、多靶点、多通路的作用特点,传统的药理学方法不能系统阐述其治疗UC的作用机制.随着生物信息学的迅速发展,网络药理学因能多系统阐述药理作用机制,与中医整体观相契合而被广泛应用于中医药治疗疾病作用机制研究[16,17].为了更好地挖掘和利用我国丰富的中医药资源,本文立足于网络药理学方法和分子对接技术系统地探讨HMR治疗UC的物质基础和作用机制.
本研究发现HMR中含有的槲皮素、豆甾醇、小檗碱、β-谷甾醇和巴马汀等为治疗UC的主要活性成分.研究表明[18],槲皮素通过诱导紧密连接蛋白修复UC模型小鼠的的肠上皮细胞,并显著降低肠道炎症损伤;豆甾醇在结肠炎中通过激活丁酸-过氧化物酶体增殖物激活受体γ轴修复调节性T细胞与辅助性T细胞17平衡,可抑制模型小鼠血液中炎症细胞增殖,有效控制结肠组织损伤[19];β-谷甾醇以浓度依赖的方式降低UC模型小鼠结肠组织中TNF-α、IL-6和IL-1β水平,并且β-谷甾醇可显著增加肠道上皮细胞抗菌肽的表达,降低致病菌(含伤寒沙门氏菌等)的存活率[20];小檗碱以微生物依赖的方式恢复葡聚糖硫酸钠诱导的UC小鼠炎症,并且小檗碱通过维持肠黏膜屏障的结构和功能、调节肠黏膜免疫稳态来发挥对结肠的保护作用[21-23];巴马汀通过抑制核苷酸结合寡聚化结构域样受体蛋白3炎症小体的活化,降低UC小鼠结肠中髓过氧化物酶、IL-1β、TNF-α的水平[24].以上结果表明从HMR中的筛选得到的多种活性成分在治疗UC方面发挥着重要的作用.
通过PPI网络拓扑分析发现,HMR治疗UC核心靶点有IL-1β、IL-6、CCL2、TNF、MMP9等.炎症反应是UC病理生理基础,炎症因子在UC肠道炎症持续过程中发挥着重要作用.研究发现,UC发病期间IL-1β分泌增加直接介导了UC初期炎症的发生,且IL-1β分泌过多可激活树突样细胞、促进白细胞介素-2受体过表达,加重结肠黏膜局部的炎症反应;IL-6分泌增加影响肠上皮细胞电解质分泌紊乱,介导其他炎症细胞发生炎性反应[26,27].TNF-α参与细胞免疫炎症反应,在UC患者受损肠黏膜中呈高水平表达,诱导结肠上皮细胞大量凋亡,从而增加肠道上皮细胞通透性[28-30].CCL2是一种重要的趋化因子[31],肠道炎症时,内皮细胞、单核细胞等细胞在TNF-α、IL-1等炎性因子刺激下会大量释放CCL2,而该趋化因子可高度特异性地激活并趋化淋巴细胞和单核-巨噬细胞等免疫细胞向正常肠黏膜组织聚集,并促进这些免疫细胞潜入发炎的肠黏膜组织,进而使肠黏膜发生更严重的免疫炎性损害[32].此外,一项临床发现[33]MMP9在活动性UC患者结肠黏膜中持续高表达,而活化的MMP9可分解多种蛋白底物,甚至导致抗TNF药物无效.近年来有学者提出,高效且高选择性的MMP抑制剂的研发或将成为UC生物制剂研发的新方向[34].上述结果提示PPI网络筛选出的核心靶点在UC的调节炎症反应和免疫应答上有着重要作用,表明HMR治疗UC主要通过上述多靶点发挥作用.
KEGG富集分析显示,HMR治疗UC主要涉及的通路为炎症反应及肠黏膜免疫相关通路,如TNF信号通路、NF-κB信号通路、Toll样受体信号通路等.本研究发现,在HMR治疗UC中TNF信号通路的基因数富集最多且表达显著.TNF信号通路可通过影响肠黏膜T细胞的活化、增殖,促进Th1细胞的极化及效应功能,上调γ干扰素的表达,从而促进肠黏膜的炎症反应[35].同时,还可作用于辅助性T细胞17使其分泌白介素17(interleukin-17,IL-17)增加,导致肠黏膜免疫失调引发肠道炎症反应.此外,TNF信号通路还能激活下游NF-κB通路,促进IL-lβ、IL-6、IL-17等炎症因子释放[36],而这些炎症因子又可以反作用于NF-κB信号通路,调控多种炎症因子相关基因,进一步加重肠道炎症反应.GO富集分析显示,HMR治疗UC与脂多糖刺激变化等生物过程有关,且多发生在细胞的质膜外侧面、膜筏、细胞外基质区域,通过影响细胞因子活性、整合素结合、丝氨酸型内肽酶活性等分子功能发挥作用.本研究通过GO、KEGG富集分析证明HMR治疗UC是多个分子、多条信号通路对多种细胞生物学行为进行调节的结果.
4 结论
综上所述,HMR可能是通过槲皮素、豆甾醇及小檗碱等多成分,作用于IL-1β、IL-6、CCL2等多靶点,调节TNF信号通路、NF-κB信号通路、TLR信号通路等多种信号通路,从抗炎和调节肠道免疫等方面发挥对UC的治疗作用.本次分析结果表明HMR治疗UC的多成分、多靶点、多通路的作用机制,为今后临床应用和基础研究提供了一定的思路和参考.
文章亮点
实验背景
溃疡性结肠炎(ulcerative colitis,UC)是一种慢性非特异性炎症性肠病,常反复发作,缠绵难愈,多数需要终身用药维持.长期使用抗UC药物可引发药物不良反应.随着中医治疗的发展,越来越多的中药方剂在治疗UC上展现出了较好的疗效.“黄连-木香-肉豆蔻”(Huanglian-Muxiang-Roudoukou,HMR)是众多名老中医治疗UC的常用组方,研究其抗UC机制有助于药物开发和推广.
实验动机
探索HMR治疗UC的作用机制,以期为该组方的进一步研究与临床应用提供参考.
实验目标
通过基因表达综合数据库(gene expression omnibus,GEO)中的基因芯片分析结合网络药理学及分子对接方法探究HMR治疗UC的潜在分子机制.
实验方法
采用GEO数据库获取UC芯片数据,运用R语言综合分析筛选出疾病差异表达基因,获得UC的疾病靶标数据库;利用中药系统药理学数据库与分析平台(traditional Chinese medicine systems pharmacology database and analysis platform,TCMSP)分别检索HMR的活性成分及其作用靶点;取疾病差异表达基因与药物作用靶点间的交集基因,通过Cytoscape软件构建“中药-活性成分-疾病-靶点”网络和蛋白互作(protein-protein interaction,PPI)网络并进行拓扑分析,以此筛选出主要活性成分及核心靶点;同时利用Metascapes数据库进行进行基因本体(gene ontology,GO)功能分析及京都基因与基因组百科全书(kyoto encyclopedia of genes and genomes,KEGG)通路;最后使用AutoDock vina软件对主要活性成分和核心靶点进行分子对接.
实验结果
共筛选得到UC差异表达基因967个,HMR活性成分29种,对应的靶点163个.“中药-活性成分-疾病-靶点”网络中有活性成分24个,主要活性成分涉及槲皮素、豆甾醇、小檗碱、β-谷甾醇、巴马汀等;蛋白互作网络中有蛋白26个,核心靶点涉及白介素1β(interleukin-1β,IL-1β)、白介素6(interleukin-6,IL-6)、CC趋化因子配体2[chemokine (C-C motif) ligand 2,CCL2]、肿瘤坏死因子(tumor necrosis factor,TNF)、基质金属蛋白酶9(matrix metallopeptidase 9,MMP9)等;GO富集分析主要涉及脂多糖刺激变化等生物过程,质膜外侧面等细胞成分,细胞因子活性等分子功能;KEGG通路分析主要涉及TNF信号通路、核转录因子κB(nuclear factor kappa-B,NF-κB)通路、Toll样受体(toll-like receptor,TLR)信号通路等;运用分子对接模拟证实排名前5的主要活性成分与核心靶点之间均具有较强的结合活性.
实验结论
通过生物信息学方法筛选及预测,HMR可能是通过槲皮素、豆甾醇及小檗碱等多成分,作用于IL-1β、IL-6、CCL2等多靶点,调节TNF信号通路、NF-κB信号通路、TLRs信号通路等多种信号通路,进而影响UC涉及的炎症、肠道免疫等多种方式,来发挥改善UC的作用.
展望前景
基于以上分析,HMR治疗UC是通过多靶点、多通路的,也提示中药的研究是较为复杂的,需要从多维度、多靶点进行研究.基于生物信息学方法,本研究初步揭示了HMR治疗UC相关靶点之间的复杂关系,也挖掘了其潜在治疗靶点及可能参与机制.但对于HMR整体治疗UC的机制仍是初步的,考虑到具体到临床中的个人更为复杂,如药物的剂量、煎煮方法、剂型等不同,会起到不一样的临床疗效.因此,基于以上不足,仍需进一步开展相关的药理学实验,进一步验证和阐明HMR治疗UC的作用机制.
猜你喜欢
中老年保健(2021年3期)2021-12-03 02:32:25
中国生殖健康(2020年7期)2020-12-10 07:48:51
材料科学与工程学报(2016年4期)2017-01-15 13:35:48
合成化学(2015年4期)2016-01-17 09:01:11
中国病理生理杂志(2015年8期)2015-12-21 12:38:06
医学研究杂志(2015年7期)2015-06-22 11:01:36
医学研究杂志(2015年3期)2015-06-10 06:41:52
创业家(2015年1期)2015-02-27 07:52:02
同位素(2014年2期)2014-04-16 04:57:16
遗传(2014年2期)2014-02-28 20:58:11
