
基于BSA分析技术对陆地棉抗旱基因的鉴定
2023-11-10 09:12孙丰磊任姣姣高文伟曲延英
新疆农业科学 2023年10期
孙丰磊,任姣姣,雷 斌,高文伟,曲延英
(1.新疆农业大学农学院,乌鲁木齐 830052;2 新疆农业科学院核技术生物研究所,乌鲁木齐 830052)
0 引 言
【研究意义】不同棉花品种的不同生长发育阶段对干旱胁迫响应不同。棉花花铃期若受到干旱胁迫会影响棉花产量。以现有棉花材料为基础,筛选抗旱性强材料,挖掘、研究棉花抗旱的相关基因,对提高棉花的抗旱性及加快培育抗旱棉花新品种的进程有重要意义。【前人研究进展】作物抗旱性是一个复杂的数量性状,传统的定位方法耗时长、成本高[1]。混池测序是一种基于二代测序以集群分离方法为主的技术,应用于基因定位和克隆等[2]。BSA(集群分离方法)应用在莴苣的抗霜霉病研究中,筛选到3个RAPD分子标记[3],3个分子标记与抗性基因Dm5/8紧密连锁[4]。通过选择具有极端性状或具有明显表型差异的亲本来构建群体,从子代群体中根据目标性状的特点选择具有极端差异子代个体,构成两个极端性状的子代DNA混池,通过测序分析获得与性状相关的分子标记,定位相关基因。由于作为两个亲本是在目标性状中选择明显差异的个体,因此,在子代的两个极端混池中,只有在目标性状基因的区域中有差异,其它区域的差异较小或基本没有差异,增加了定位的准确性和可靠性。混池测序(BSA-seq)早期应用在基因组较小的物种中,并且目标基因在该物种的突变体中。Schneeberger等[5]利用混池测序成功定位到导致拟南芥叶片浅绿色突变体的基因。在拟南芥中还定位到配子突变导致不育的基因[6]以及突变导致开花延迟的基因[7]。而此方法在水稻中也得到应用,Abe等[8]在EMS诱变的突变体水稻中将白化和半矮秆定位在某一区间内,进而对目标进行了定位和克隆。Takagi等[9]在EMS诱变的水稻突变体中,定位到一个耐盐碱基因,通过MutMap 的方法克隆该基因并培育出耐盐碱的品种。陈忠明等[10]结合BSA分析法和RFLP的方法,在可育与半不育的极端混池中发现了2个标记(RG213、G200)与亲和基因连锁。陆光远等[11]利用油菜的回交群体筛选到两个AFLP标记,2个标记与细胞核雄性不育基因紧密连锁。李传友等[12]结合BSA和AFLP分析法筛选到光敏核不育基因相关的多态性产物。张显等[13]在西瓜隐形和雄性不育和可育的植株中,通过RAPD-BSA方法筛选出s1167的引物可以区分该两种株系。Wang等[14]通过BSA分析的方法在水稻中筛选到9个分子标记,利用9个分子标记最终定位到tms5基因。Geng等[15]利用BSA分析法对油菜千粒重基因进行定位,筛选到4个与千粒重相关的候选基因。Hong等[16]通过该方法筛选到与甜味相关的候选基因1个,酸味相关的候选基因8个。Zheng等[17]通过混池测序,共筛选到4个抗稻瘟病候选基因。而Zhang等[18]通过混池关联分析的方法研究黄瓜抗白粉病相关基因,共筛选到6个相关的候选基因。Chen等[19]和Feng等[20]通过BSA测序分析,再结合SSR标记,定位到与抗稻瘟病相关的基因。Qin等[21]在大麦白化性状方面利用集群分离分析的方法,结合SSR分子标记,定位6个有关该性状的候选基因。杜威世等[22]在海岛棉品种和陆地棉品种的杂交F2代中,结合SSR分子标记和BSA法定位到3个与抗棉花黄萎病相关的基因标记。Hayes等[23]研究结果表明,在大豆中通过BSA法筛选到4个Rsv1相关的分子标记,该4个分子标记与抗花叶病相关。【本研究切入点】棉花抗旱性是一个复杂的数量性状,传统的定位方法耗时长、成本高。混池测序是一种基于二代测序以集群分离方法为主的技术,可以有效用于棉花复杂性状的遗传定位。【拟解决的关键问题】筛选出抗旱性强和抗旱性敏感的材料,构建抗旱性强和抗旱性弱的两个极端样本混池,采用BSA混池测序技术测序分析亲本及子代混池,定位筛选抗旱基因位点候选区域,为挖掘棉花相关的抗旱基因奠定基础。
1 材料与方法
1.1 材 料
选用由石远321为父本和奎85-174为母本构建的150份材料(重组自交系子代群体2年2点田间抗旱表型鉴定),筛选得到抗旱性强的20份极端材料和20份水分敏感的极端材料,分别提取极端材料的叶片DNA,检测浓度以及纯度,等量混合,分别构成极端抗旱池与极端敏旱池,提取2个亲本的叶片DNA。
样本混池建库和测序由北京百迈客生物科技有限公司完成。按照lllumina公司执行标准,首先检测DNA样品后,进行超声破碎[24],修复、拼接、扩增,并构建完成测序所需要的文库。经过质检合格后的文库用测序分析(参考陆地棉基因组(版本号为v2.1))。
1.2 方 法
1.2.1 SNP变异位点的检测
SNP变异位点检测是通过GATK[25-26]软件工具包完成。根据Clean Reads在参考基因组的定位结果,通过去重复(Mark Duplicates)以及GATK进行局部重比对(Local Realignment)、碱基质量值校正(Base Recalibration)等处理,再使用GATK检测单核苷酸多态性(Single Nucleotide Polymorphism,SNP),得到SNP位点集。
根据Clean Reads在参考基因组上的定位结果,小片段的插入与缺失有基因组之间的检测确定。但同样反映了其差异的Small InDel变异的数量较少,其中InDel时会引起移码突变[27],如果移码突变区在编码区将导致基因功能上的改变。
1.2.2 关联分析及候选区域的基因注释
对SNP过滤后,进行关联分析。在SNP关联分析中主要采用欧式距离(Euclidean Distance,ED)和SNP-inde 2种分析方法,将2种分析方法共同关联到的区间作为抗旱相关的主要区间。
其中欧氏距离分析方法(Euclidean Distance,ED)是通过分析测序数据在混池间确定差异显著的标记,确定与目标性状关联的区域。在目标性状相关位点上除了2个子代混池与亲本间存在显著差异外,其它位点不存在差异或趋于基本一致,通过分析混池间差异的SNP位点,计算ED值,在实验中取ED的2次方进行拟合。
1.2.3 荧光定量验证候选基因的差异表达
将10份抗旱性极端材料在实验室内进行土培(土壤为黑土∶蛭石为2∶1混合)培养至三叶期时,进行土壤水分胁迫试验,用于荧光定量测定基因表达量。
1.3 数据处理
统计混池间基因型频率差异的分析方法应用在标记关联分析中称为SNP-index算法[28]。是通过Δ(SNP-index)统计分析混池间基因型频率间的显著差异。选择性状相关的区域是在标记的染色体上通过ΔSNP-index值进行拟合分析后关联阈值以上的区域。
通过BLAST软件对候选基因在多个数据中(GO[20]、KEGG[21]、NR[22])比对详细注释。
2 结果与分析
2.1 样品测序数据对比
研究表明,测序得到原始reads为119 060 015、121 194 054、190 811 397、223 420 448,得到clean reads与原始reads基本一致,总数据量为197.65 Gbp,其中Q30(%)在93%以上,并且GC含量在35.43%~35.73%,共得到142.23 Gbp的Clean reads,平均每个样品测序深度15.47X,最终的平均比对效率为99.045%,达到18.75X平均覆盖深度和99.03%的基因组覆盖度。表1,表2
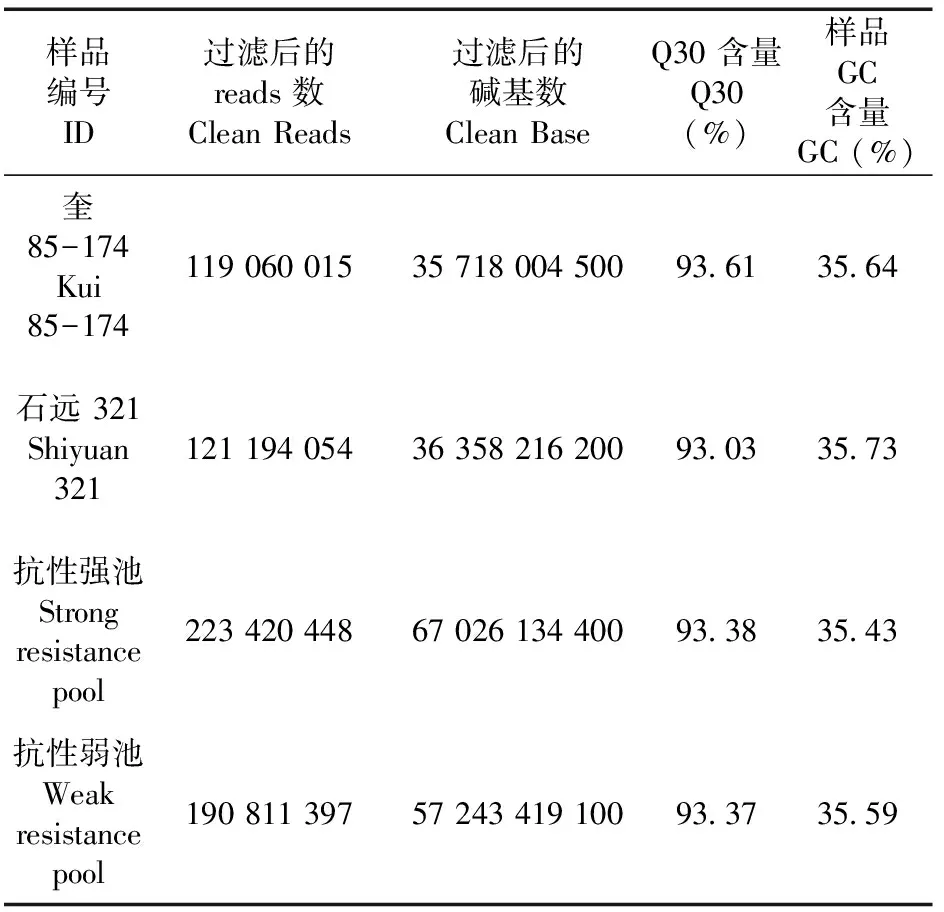
表1 样品测序数据质量统计

表2 与参考基因比对
2.2 变异位点检测
研究表明,亲本之间共获得1 145 784个SNP,其中非同义突变的SNP共13 753个;混池之间共获得588 221个SNP,其中引起非同义突变的SNP共6 186个。亲本之间共获得221 481个Small InDel;混池之间共获得134 957个Small InDel。图1
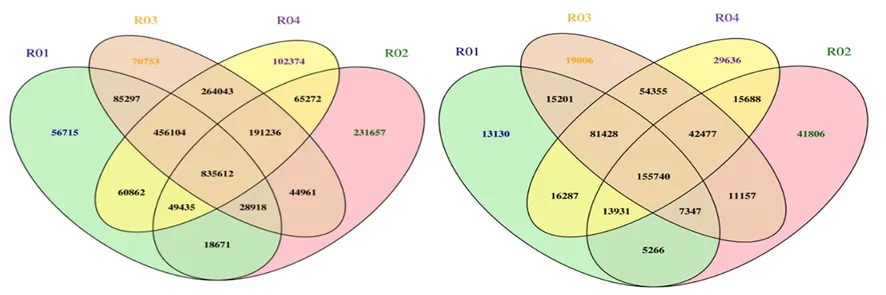
图1 样品间SNP(左)和InDel(右)Venn图
2.3 抗旱性关联
2.3.1 SNP检测
研究表明,共得到2 561 910个SNP位点,再次过滤后最终得到高质量SNP位点共540 596个。
取所有位点的拟合值作为关联阈值,得到ED值为0.17,共得到10个区域,总长度为39.30 Mb,共包含979个基因,其中包含非同义突变SNP位点的基因共112个。图2,表3

注:横坐标为染色体名称,彩色的点代表每个SNP位点的ED值,黑色的线为拟合后的ED值,红色的虚线代表显著性关联阈值,下同

表3 ED算法关联候选区域
拟合后的ΔSNP-index值为0.31,共得到5个区域,总长度为6.12 Mb,共包含86个基因,在所有基因非同义突变的共有27个。图3,表4

图3 ΔSNP-index关联值在染色体上分布

表4 SNP-index算法关联候选区域
2.3.2 候选区域筛选及候选区域基因注释
研究表明,共筛选到3个候选区域,总长度为6.12 Mb,共包含86个基因。表5
表5 ED和SNP-index两种算法关联候选区域
亲本间存在非同义突变的SNP共43个,混池间存在非同义突变的SNP共20个,在候选区域内共注释到83个基因,而注释到的非同义突变基因在亲本间共有27个。其中在GO数据库中注释到49个基因,而在KEGG数据库中注释到23个基因。
候选区域内每个组分中基因数量不同,其中大多数被划分到生物过程(Biological process)中。前5的功能分类分别是:在细胞组分中为质体部分、叶绿体部分、细胞器膜、转移酶复合体和叶绿体;在分子功能中为RNA导向的DNA聚合酶活性、核糖二磷酸羧化酶活性、蛋白质结构域特异性结合、双加氧酶活性、碳水化合物磷酸酶活性;生物过程中主要为DNA整合、依赖RNA的DNA复制、糖醇代谢过程、细胞壁组织或生物合成、有机羟基化合物代谢过程。图4,表6

图4 候选区域内基因GO注释聚类
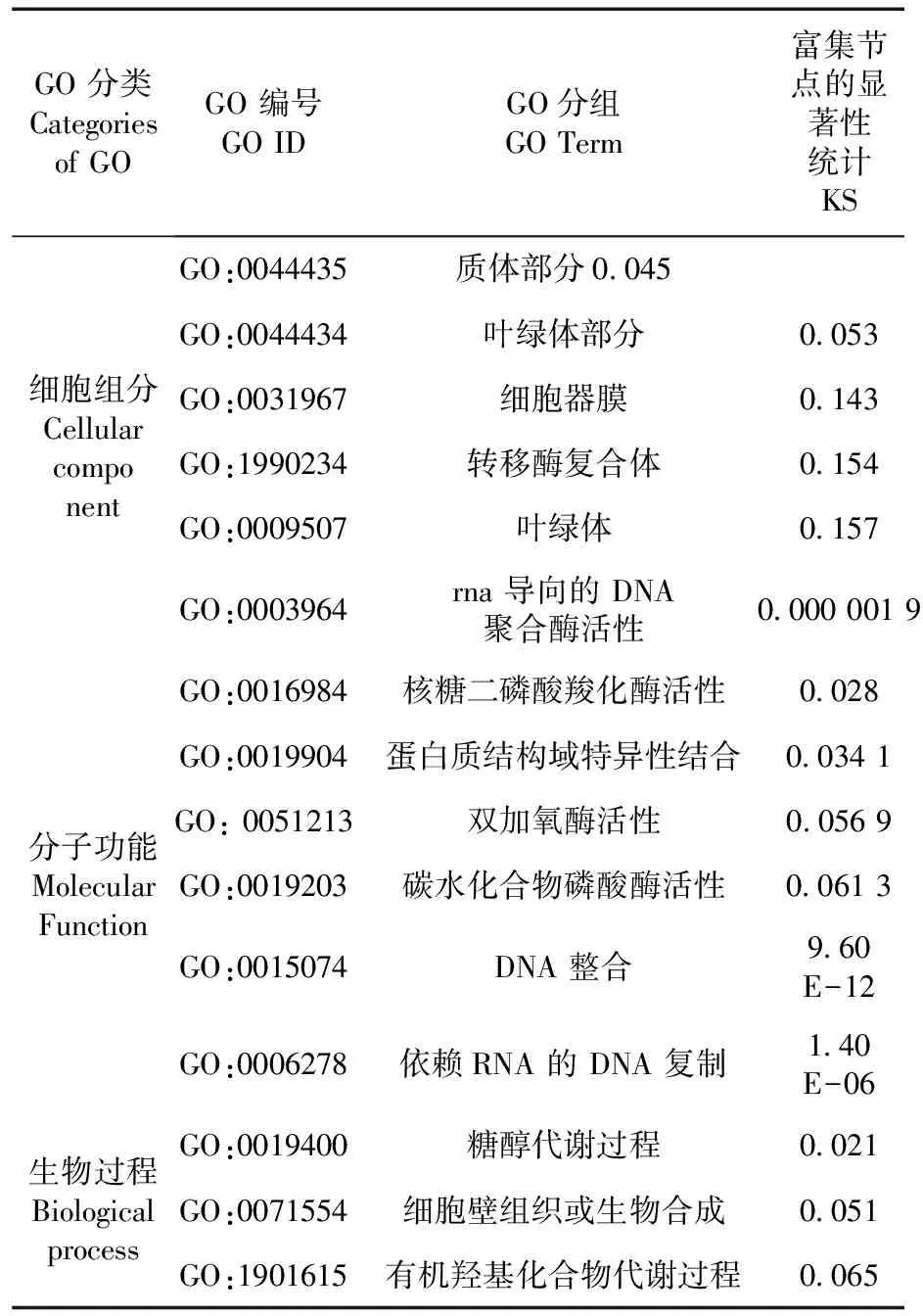
表6 候选区域内基因GO注释
得到的代谢通路中基因注释分布最多的在代谢作用和遗传信息处理中,代谢作用通路中注释基因最多的是内质网中的蛋白质加工和RNA聚合酶;在遗传信息处理的代谢通路上苯丙烷生物合成和氨基酸生物合成中注释到的基因种类最多。图5
2.3.3 候选基因的筛选
研究表明,参与细胞组成的相关基因与棉花的抗旱性相关,包括细胞核(GO:0005634),叶绿体(GO:0009507),过氧化物酶体(GO:0005777),RNA聚合酶Ⅲ复合物(GO:0005666)等,GH_A07G0705、GH_A07G0701、GH_D07G1930、GH_D07G1932、GH_D07G1934、GH_A07G0708、GH_A07G0695、GH_A07G0703等8个基因参与调控棉花的抗旱性。
锌离子结合(GO:0008270)、RNA定向的DNA聚合酶活性(GO:0003964)、蛋白质结构域特异性结合(GO:0019904)、甲基转移酶活性(GO:0008168)等参与到干旱胁迫相关反应中。GH_A07G0695、GH_A10G1602、GH_D07G1927、GH_A10G1623、GH_A10G1601等24个基因与干旱胁迫调控相关。
一些参与干旱胁迫调控的过程,如代谢过程(GO:0008152),响应脱落酸反应过程(GO:0009737),有机物的运输过程(GO:0071702),还有氧化还原过程(GO:0055114)等均参与干旱胁迫的生物调控过程中。预测GH_A07G0696、GH_A07G0700、GH_A07G0695、GH_A07G0703、GH_A07G0695、GH_A07G0696、GH_A07G0705等24个基因与干旱胁迫相关。表7
筛选出与前期5个棉花抗旱性评价关键性状指标相关的15个候选基因,分布在3个候选区域内。表8

表8 抗旱候选基因及注释
2.4 qRT-PCR分析候选基因表模式
研究表明,在水分胁迫达到轻度(土壤含水下降了20%)、中度(土壤含水下降了40%)、重度(土壤含水下降了50%)时,在胁迫程度增加时,GhalkB随着胁迫程度的增加都有一个上调表达的趋势,在石远321中该基因在土壤含水量下降了10% 时表达量最高,随着胁迫程度的增加,该基因的表达量先下降后又上升。而奎85-174在下降了10%时最高。在测定的时期中,该基因在石远321中表达量始终高于奎85-174。
GhGMPPA同样是随着胁迫程度的增加,呈现一个上调表达的趋势,在石远321中该基因在土壤含水量下降了40% 时表达量最高。而奎85-174在下降了10%时 最高,两个阶段中抗旱性强的材料中的表达量显著高于抗性弱的材料。GhPPT1在亲本中表达情况是随着时间的增加都有一个上调表达的趋势,在石远321中该基因在土壤含水量下降了10% 时表达量最高,随着胁迫程度的增加,表达量呈下降趋势。而奎85-174在下降了10%时最高。而GhASP1在受到干旱胁迫后,随这胁迫程度增加时,该基因呈现一个上调表达的趋势,石远321中该基因在土壤含水量下降了50% 时表达量最高,随着胁迫程度的增加,表达量呈上升趋势。而在奎85-174中则在下降了10%时最高。GhRBCS与GhGMPPA相同,同样是在远321中该基因在土壤含水量下降了40% 时表达量最高。而奎85-174在下降了10%时最高。并且在所有的测定时期中,5个基因在石远321中表达量始终高于奎85-174。
在受到水分胁迫时,抗旱性强的材料中这些基因的表达量均有上调表达的趋势,并且抗旱性强的材料在轻度和中度水分胁迫的条件下的5个基因表达量显著高于抗旱性弱的材料。图6

注(Note):a:GhalkB,b:GhGMPPA,c:GhPPT1,d:GhASP1,e:GhRBCS
在干旱胁迫第3 d时,GhalkB在抗旱性强的材料中表达量显著高于抗旱性弱的材料,并且在此时GhalkB的基因表达量达到最大值。由于呈快速上调表达的趋势,该基因对干旱胁迫前期较为敏感。而在抗旱性弱的材料中表达量变化不显著。GhalkB在干旱胁迫前期起着重要作用,抗旱性强的材料在轻度胁迫时表现高表达,抗旱性弱的材料表达量变化不显著,该基因可能与棉花抗旱性有关。胁迫第3 d时,GhGMPPA在抗旱性强的材料中表达量显著高于抗旱性弱的材料,也在此时达到最大值。受到胁迫后GhGMPPA基因呈现一个快速上调的表达趋势,但是随着胁迫程度的增加,呈现一个逐步下降的过程,而在抗旱性弱的材料中表达量的变化不显著。抗旱性强的材料中新陆早45号、贝尔斯诺、石远321三个材料在胁迫3 d时表达量达到最高,并且显著高于抗旱性弱的材料,而ND359-5和KK153是在胁迫第6 d时表达量达到最大,并且显著高于抗旱性弱的材料,该基因在不同材料中对干旱胁迫程度的敏感度不同;而在抗旱弱的材料中变化不显著。同样是在干旱胁迫第3 d(达到轻度胁迫条件)时,GhASP1基因表达量最高,并且显著高于抗旱性弱的材料,也呈现快速上调表达的趋势,在抗旱弱的材料中表达量变化不显著。在干旱胁迫第6 d时(中度水分胁迫),在抗旱性材料中表达量达到最大,并且显著高于抗旱性弱的材料,其中新陆早45号、贝尔斯诺在胁迫第3 d时的表达量也高于其它材料,呈现一个逐步上升的趋势,该基因在不同材料中对干旱胁迫程度的敏感度不同;而在抗旱弱的材料中变化不显著。图7

注(Note):a:GhalkB,b:GhGMPPA,c:GhPPT1,d:GhASP1,e:GhRBCS
3 讨 论
3.1Schneeberger等[5]利用混池测序在拟南芥中成功定位到导致叶片浅绿色突变体的基因。同时在拟南芥中还通过混池测序定位到配子突变导致不育的基因[6],以及突变导致延迟的基因[7]。Han等[29]在高粱茎含水量性状相关研究汇总通过BSA测序分析定位关联到6号染色体上339kb的区间内[30]。贾秀萍等[31]通过高耐盐自交系Y07-136R(父)和耐盐敏感材聊Y05-222A(母)构建的F2群体,通过BSA测序共发掘到6个耐盐关键候选基因。刘梦雨等[32]用多油泡的罗纹金柑和少油泡的滑皮金柑杂交构建F1群体,通过重测序-BSA分析共获得了11个相关的候选基因。张尧峰等[33]在油菜中以有限花序突变株系FM8与野生型自交系 FM7杂交,F2中挑选子代构建混池,获得3个候选基因。研究中,以石远321为父本、奎85-174为母本构建了陆地棉重组自交系。分别以重组自交系群体子代中,20份抗旱性强的子代材料和20份抗旱性弱的子代群体材料构建混池,通过BSA测序分析,在SNP-index和ED两种算法进行关联分析后取交集,最终关联到3个相关的候选区域,候选区域的总长度为6.12Mb,其中共包含86个基因。结合GO注释和KEGG通路分析,该基因在分子功能、细胞组分、或生物过程中有参与到干旱调控的过程中,参与了棉花的抗旱响应过程。
3.2研究通过抗旱性评价筛选出抗旱性极端材料,结合BSA测序分析,筛选抗旱相关候选基因。通过测序数据分析及生物信息学分析,结合KEGG通路分析及基因注释相关信息,共筛选出15个抗旱相关的候选基因,亲本以及极端资源材料中荧光定量的测定中显示,在不同程度的胁迫中,5个基因在亲本和极端材料中表现出不同差异,其中alkB、GMPPA、PPT1、ASP1、RBCS基因的表达量在抗性强的材料中显著高于抗旱性弱的材料,并且在前期抗旱性评价筛选出的5个关键指标性状的相关通路中,确定alkB、GMPPA、PPT1、ASP1、RBCS5个候选基因。通过数据库比对分析,alkB是一种双加氧酶通过影响水分运输,导致叶片失水影响叶片的生长发育;GMPPA是GMPPB的同源物,可催化GDP-甘露糖的形成,后者是糖蛋白和糖脂的糖基部分的重要前体;PPT1作为磷酸烯醇丙酮酸转运酶,将磷酸烯醇丙酮酸作为草酰乙酸途径的一种底物导入叶绿体基质中,同时参与叶肉细胞发育;ASP1作为氨基酸氨基转移酶,参与氨基酸和三羧酸循环,同时参与氮代谢及碳代谢方面;RBCS主要是在二氧化碳固定中将戊糖等进行氧化裂解,参与光合作用。
4 结 论
得到3个关联区域,总长度为6.12Mb,共包含86个基因。结合qRT-PCR分析,共筛选出GhalkB,GhGMPPA,GhPPT1,GhASP1,GhRBCS5个基因,以石远321正常对照cDNA为材料克隆了5个基因编码序列,除GhPPT1存在跨膜区域,属于膜蛋白外,其余基因均不存在跨膜区域,因此不属于膜蛋白;其中GhRBCS亚细胞定位预测结果显示定位于叶绿体中,预测定位位于线粒体中,而GhGMPPA和GhalkB则定位预测在细胞质中,而GhPPT1则预测定位于叶绿体膜上;GhalkB基因含有Fe(2+)2-氧戊二酸双加氧酶结构域,与亚洲棉亲缘关系最近;GhGMPPA基因在N末端域是GDP-甘露糖焦磷酸化酶同工型结构域,其与雷蒙德氏棉亲缘关系最近;GhPPT1基因具有磷酸三糖转运蛋白家族蛋白结构域,也与亚洲棉亲缘关系最近;GhASP1基因具有PLP依赖性酶的天冬氨酸转氨酶(AAT)结构域,也与亚洲棉亲缘关系最近;GhRBCS基因含有APF序列的保守结构域,其中亲缘关系最近的是海岛棉和陆地棉的。在根、茎、叶中均有表达,但是该5个基因在叶片中显著表达。
猜你喜欢
水利水电快报(2023年2期)2023-02-21
今日农业(2021年14期)2021-11-25
今日农业(2020年13期)2020-12-15
世界科学技术-中医药现代化(2020年2期)2020-07-25
河南水利年鉴(2017年0期)2017-05-19
浙江农业学报(2017年1期)2017-05-17
临床医药文献杂志(电子版)(2017年11期)2017-05-17
西南农业学报(2016年4期)2016-05-17
中学生物学(2016年8期)2016-01-18
山西农经(2015年7期)2015-07-10
