
pH和温度双响应性的微凝胶复合膜的制备及油水分离性能研究
2023-11-07 06:43:04陈富有赵雅雯高从堦陆叶强薛立新
膜科学与技术 2023年5期
陈富有, 赵雅雯, 马 慧, 高从堦, 陆叶强*, 薛立新,2*
(1. 浙江工业大学 化工学院 膜分离与水科学技术中心, 杭州 310014;2. 温州大学 化学与材料工程学院, 温州 325035)
由于全球经济增长、快速工业化和人口增长,水污染已成为日益严峻的环境挑战[1-2].含油废水被认为是一种主要的水污染,通常由化工生产、石油泄漏、车辆运输和生活污水产生,大量的排放对生态环境造成了巨大的影响[3-5].传统方法,包括原位燃烧、重力沉降、离心、吸附、浮选和生物方法,已被用于处理含油废水[6-11].然而,这些方法通常伴随着一定的缺点,如能耗高、分离效率低、操作复杂、存在二次污染等,极大地限制了它们的应用[12-13].因此,迫切需要开发节能、高效、环保、分离性能优异和多功能的先进油水分离策略.
近年来,开发特殊浸润性材料已成为材料研究的热门话题之一.特别的,对油和水具有明显相反亲和力的特殊浸润性膜材料被认为是选择性油/水分离最有前途的材料[14-16].浸润性是固体材料表面的一种固有属性,由材料的表面形态和化学成分决定.受自然界中荷叶、玫瑰花瓣和鱼鳞的启发,研究人员发现,通过调控材料的表面自由能,构建微纳米粗糙结构,膜材料可以被赋予极高的浸润性[17-19].于是,研究人员基于膜表面自由能和表面微纳结构的同步调控,设计并制备了一系列具有不同表面浸润性的膜材料,如超疏水/超亲油膜和超亲水/超疏油膜等.超疏水/超亲油膜可以限制水的通过,同时有利于油的通过,可选择性地从油水混合物中分离油[20].例如,Sun等[21]通过静电纺丝技术制备了一种超疏水/超亲油碳纤维膜,仅在重力驱动下可用于分离油包水乳液.超亲水/超疏油膜允许水的通过而限制油的通过,可选择性地从油水混合物中分离水[22].例如,Yang等[23]将二氧化硅纳米颗粒与聚二甲基氯化铵和全氟辛酸钠配位合成的聚合物黏合剂混合溶液喷涂在不锈钢网膜上,得到一种超亲水/超疏油膜,可在重力驱动下实现对油水乳液的分离.上述材料都可以成功地实现大多数油/水混合物的分离.然而它们的表面浸润性单一,通常具有各种局限性(如油水选择性差、易污染和孔隙率低等),对于体系复杂的含油废水,并不能起到一个有效的分离[24-26].因此,急需开发出具有可切换浸润性的刺激响应膜材料.
刺激响应膜,也称为智能膜,由刺激响应材料制成,能快速应对外界环境刺激并作出相应反应,调控膜表面的浸润性能,完成从单一浸润性到多重浸润性的转变,从而实现对不同种类的油水混合物进行高效分离.例如,Chen等[27]通过接枝光响应偶氮苯同步调控微纳米粗糙结构的方法,成功制备了智能光响应聚丙烯膜,通过控制紫外光/可见光的交替照明,从而实现在“除油”和“除水”状态之间的可逆转换.然而,制造工艺复杂,一些试剂相对昂贵,极大地限制了其实际应用.
本研究首先通过简单的自由基聚合法制备了具有pH和温度双刺激响应的含氟P-PDA微凝胶.然后以PVDF为基膜材料,通过共混P-PDA微凝胶的方式得到改性铸膜液,接着采用乙醇作为凝固浴通过相转化的方式制备出了一种具有pH和温度双刺激响应性的微凝胶复合膜.采用乙醇作为凝固浴延缓了膜相转化所需要的时间,改善了微凝胶对凝固浴的亲和力,促使更多的微凝胶迁移至膜表面.这些微凝胶一方面有利于膜表面自由能和膜微纳粗糙结构的同步调控,另一方面可以充当膜的刺激响应开关,进而影响膜表面的浸润性.制备的膜在空气中表现出高疏水性(147.2°)和超亲油性(0°),当膜被pH=13水预处理后,空气中的高疏水性和超亲油性转变为亲水性和水下超疏油性.由于其具有可切换的浸润性,仅在重力驱动下成功实现了对W/O和O/W乳液的有效分离,且具有优异的自清洁性能.
1 实验部分
1.1 实验材料
PVDF(Kynar 761A),法国阿科玛有限公司;N-异丙基丙烯酰胺(NIPAM),α-甲基丙烯酸(MAA),甲基丙烯酸三氟乙酯(FMA),2,2-偶氮二异丁腈(AIBN),吐温80,司班80,阿拉丁(上海)试剂有限公司;盐酸多巴胺(DA),γ-甲基丙烯酰氧基丙基三甲氧基硅烷(MPS),N-甲基吡咯烷酮(NMP),甲苯,上海泰坦科技股份有限公司;无水乙醇,氨水(28%~30%),正己烷,石油醚,液体石蜡,泵油,苏丹Ⅲ,氢氧化钠(NaOH),国药集团化学试剂有限公司.
1.2 P-PDA微凝胶的制备
将氨水溶液(NH4OH,1.5 mL,28%~30%)与去离子水(180 mL)和无水乙醇(80 mL)在室温下温和搅拌混合30 min.将DA(1.0 g)溶解于去离子水(20 mL)中,然后注入上述混合溶液中.该溶液的颜色立即变为浅棕色并逐渐变为深棕色.允许反应进行24 h后,将MPS(2.5 g)加入上述混合物溶液中,在60 ℃下连续搅拌8 h.离心(11 000 r/min、10 min)、洗涤(3次、无水乙醇)、干燥,得到粒径为450 nm的双键改性的PDA-MPS纳米粒子.然后,合成了具有pH和温度双刺激响应的含氟P-PDA微凝胶.通常,将PDA-MPS(0.3 g)分散于无水乙醇(60 mL)中,超声20 min使其分散均匀.然后将FMA(1.008 g,6 mmol)、NIPAM(0.678 g,6 mmol)和MAA(0.689 g,8 mmol)加入上述溶液中,氮气除氧30 min,并放入60 ℃油浴锅中.向溶液中加入AIBN(0.0656 g,2 mmol)反应8 h.所有过程均在氮气下搅拌.离心(11 000 r/min、10 min)、洗涤(3次、无水乙醇)、干燥,得到P-PDA微凝胶.
1.3 P-PDA微凝胶复合膜的制备
将PVDF粉末(4.0 g)添加到50 mL圆底烧瓶中的NMP(18.2 g)中.然后将混合物在70 ℃下搅拌溶解,形成透明的溶液.随后,将溶解于NMP(3.0 g)中的P-PDA微凝胶(0.5 g)添加到该溶液中,并将混合物在70 ℃下搅拌溶解,形成均一的铸膜液.最后,将混合物静置过夜以除去铸膜液中的气泡.将上述制备的铸膜液静置脱泡后,在环境温度23±1 ℃、湿度38%±2%下刮膜,膜厚为300 μm.将平板膜浸入乙醇凝固浴中相转化15 min,以诱导P-PDA微凝胶向膜表面迁移.最后将平板膜置于去离子水中清洗,取出于25 ℃下干燥,得到具有温度和pH双刺激响应性的P-PDA微凝胶复合膜,并命名为M2.同时用纯水凝固浴制备了纯PVDF膜(M0)和P-PDA微凝胶改性PVDF膜(M1)用于比较.
1.4 仪器与表征
通过扫描电子显微镜(SEM, HITACHI,SU 8010,Japan)和透射电子显微镜(TEM,TEM-low Lx Ⅱ, Japan)观察并分析了微凝胶和膜的表面形貌.采用动态光散射粒径分析仪(DLS,90 Plus,Brookhaven Instruments Corporation,USA)测试了微凝胶的流体动力学直径.借助原子力显微镜(AFM,Bruker Dimension Edge,USA)分析膜表面形貌和粗糙度.以KBr为空白,采用傅里叶变换红外光谱仪(Thermo Nicolet 6700,USA)测试膜在550~4 000 cm-1的傅里叶红外光谱.通过能量色散光谱(EDS)和X射线光电子能谱(XPS,Thermo ESCALAB 250 XI,USA)测量了膜表面的元素分布和含量.通过接触角测量仪(DataPhysics,OCA 20,Germany)测试膜的水和油接触角及水下油接触角,测试液滴体积为3 μL,每个样品均测试5个点,取其平均值.通过流动法毛细管孔径分析仪(Porometer,Porolux 500,Germany)测试了膜的孔径分布.通过偏振光显微镜(BX53M,Olympus,Japan)观察油/水乳液分离前后的液滴尺寸分布.分别用总有机碳分析仪(TOC-LCPH,Japan)和卡尔费休水分测定仪(AKD-A 3,Arkray Instrument,China)测试了乳液在分离前后溶液中有机物和水含量的变化.
1.5 油水乳液的制备及其性能评价
表面活性剂稳定的W/O乳液的制备:将油(使用正己烷、石油醚、液体石蜡和甲苯)和水以95∶5的体积比混合,加入1.2 mg/mL的司班80,在磁力搅拌下持续搅拌4 h,直至溶液变成均匀的乳白色.使用相同的方法通过混合含有1.2 mg/mL吐温80的水和油(95∶5)来制备O/W乳液.所制备的乳液可在室温下保持稳定超过24 h且无明显分层.
在室温下,仅在重力条件下,在死端过滤设备上进行油水乳液分离实验.将所制备的膜放置在两个玻璃容器之间,有效膜面积为13.85 cm2.渗透率(F)和分离效率(R)分别通过式(1)和式(2)计算.
(1)
(2)
式中:V是滤液体积,L;A是膜有效面积,m2;T是分离时间,h;C和C0分别是分离前乳液和分离后滤液中相应水或油的浓度,g/L.
2 结果与讨论
2.1 P-PDA微凝胶的形貌和双刺激响应行为
为了验证P-PDA微凝胶的成功合成,使用SEM和TEM对最终产物的核壳结构进行表征.如图1所示,通过SEM可以观察到,纯PDA微球具有完美且规则的球形结构,平均尺寸为500 nm左右,表面光滑且分散性良好[图1(a)~1(c)].相比之下,P-PDA微凝胶具有粗糙的表面[图1(d)~1(f)].表面形貌的变化归因于聚合物壳在PDA微球表面的成功覆盖,复合微球比由单一材料组成的微球更有可能具有粗糙的表面形态[28].此外,TEM图像生动地显示了PDA微球的规则球形结构[图1(g)~1(h)]和以PDA微球为核心,以聚合物为外壳的P-PDA微凝胶的典型核壳结构[图1(i)].可以观察到,厚度为30 nm的聚合物壳很好地包围了PDA微球.以上结果证明了P-PDA微凝胶的成功合成.

图1 (a~c)纯PDA和(d~f)P-PDA微凝胶的SEM图像,(g~h)纯PDA和(i)P-PDA微凝胶的TEM图像Fig.1 SEM images of (a~c) pure PDA and (d~f) P-PDA microgels, TEM images of (g~h) pure PDA and (i) P-PDA microgels
P-PDA微凝胶的pH和温度响应性能通过DLS进行表征.首先将所制备的微凝胶超声使其均匀分散在去离子水中.微凝胶的浓度为0.05 g/L.随后将微凝胶分散液的温度/pH分别调节至25 ℃/pH=13、25 ℃/pH=7和45 ℃/pH=7.然后,通过DLS测量微凝胶在上述条件下的粒径分布.如图2(a)~2(c)所示,P-PDA微凝胶的流体动力学直径从25 ℃/pH=13时的645 nm降低到45 ℃/pH=7时的561 nm.微凝胶的刺激响应行为由pH响应聚合物PMAA和温度响应聚合物PNIPAM共同控制.在25 ℃下,当分散液的pH从pH=13调节至pH=7时,由于质子化的羧基和显著的疏水相互作用,P-PDA微凝胶显示出体积收缩[26].进一步,当分散液的温度从25 ℃升高至45 ℃时,高于PNIPAM的LCST≈32℃.此时,PNIPAM中的酰胺基团之间优先形成分子内氢键,疏水链段起主导作用,表现出疏水性,从而导致P-PDA微凝胶的粒径进一步缩小[29].

(a)25 ℃/pH=13,(b)25 ℃/pH=7,(c)45 ℃/pH=7图2 P-PDA微凝胶在不同pH/温度下的粒径分布Fig.2 Particle size distribution of P-PDA microgels at different pH/temperatures
2.2 膜表面及断面形貌结构分析
为了研究P-PDA微凝胶及乙醇凝固浴对PVDF膜性能的影响,使用SEM表征所得膜的表面及断面形貌.如图3(a)、3(a′)和3(a″)所示,通过采用水凝固浴诱导相分离(NIPS)获得的M0膜具有致密的表面.添加P-PDA微凝胶后,M1膜表面的孔径较M0膜并未有明显的改变,只是在M1膜表面能够观察到零星的几个微凝胶[图3(b)、3(b′)和3(b″)].这是由于聚合物壳中含氟链段的存在,导致P-PDA微凝胶呈现一定的疏水性,由于疏水相互作用,导致P-PDA微凝胶不利于向膜表面迁移.当凝固浴由水换成乙醇后,M2膜表面形貌发生显著变化.如图3(c)、3(c′)和3(c″)所示,M2膜表面显得更为粗糙多孔且在膜表面还镶嵌着大量的微凝胶.这种显著的形态变化可归因于NIPS工艺的变化:一方面,采用乙醇作为凝固浴削弱了铸膜液与凝固浴之间扩散传质交换的化学势变化,从而降低了溶剂与非溶剂在相转化过程中的扩散速率[30].另一方面,与PVDF在水中的溶解度相比,PVDF在乙醇中的溶解度更小,沉淀速度更慢[31].上述因素都有利于膜大孔结构的生成.此外,采用乙醇作为凝固浴延缓了膜相转化所需要的时间,改善了微凝胶对凝固浴的亲和力,促使更多的微凝胶迁移至膜表面[32].图4展示了M0膜、M1膜和M2膜的断面形貌.如图4(a)、4(a′)和4(a″)所示,M0膜存在海绵状的多孔结构,孔道表面光滑.添加P-PDA微凝胶后,M1膜断面形貌并未发生明显变化,只是在M1膜孔道周围能够观察到少量的的微凝胶[图4(b)、4(b′)和4(b″)].当凝固浴由水换成乙醇后,M2膜断面形貌发生显著变化且在膜孔道周围分布着更多的微凝胶[图4(c)、4(c′)和4(c″)].该结果与表面形貌结果相一致.

图3 不同放大倍率下膜表面的SEM图像Fig.3 SEM images of the surface of the membranes at different magnification

图4 不同放大倍率下膜断面的SEM图像Fig.4 SEM images of the cross-section of the membranes at different magnification
为了进一步探究P-PDA微凝胶及乙醇凝固浴对PVDF膜表面形貌的影响,使用AFM分析膜表面的粗糙度.如图5(a)所示,M0膜的算术平均粗糙度(Ra)为28.3 nm.添加P-PDA微凝胶后,M1膜的算术平均粗糙度略有增加,达到了35.2 nm[图5(b)].将凝固浴由水换为乙醇后,M2膜的算术平均粗糙度明显增加,达到了116.5 nm[图5(c)].这可以从两个方面来解释:一方面,凝固浴乙醇的存在有助于增加膜表面不平整度和粗糙度[30].另一方面,采用乙醇作为凝固浴延缓了膜相转化所需要的时间,有利于P-PDA微凝胶向膜表面迁移,进一步增加了膜表面的粗糙度.该结果与上述SEM非常吻合.

图5 (a)M0膜、(b)M1膜和(c)M2膜的AFM图像Fig.5 AFM images of (a) M0, (b) M1 and (c) M2 membranes
2.3 膜表面的化学结构和成分分析

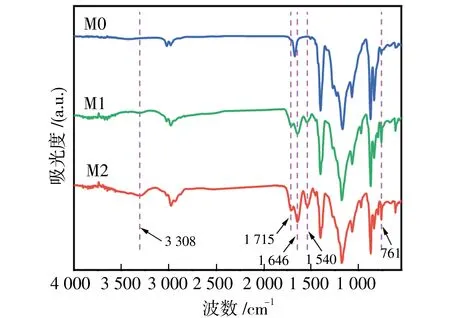
图6 M0膜、M1膜和M2膜的FTIR光谱Fig.6 FTIR spectra of M0, M1 and M2 membranes
此外,还进行了高分辨率XPS以及EDS表征,以研究膜表面原子组成.XPS谱图如图7所示,M0膜表面仅在283.0和685.0 eV出现碳(C 1s)和氟(F 1s)的两个峰.附着微凝胶后,M1膜表面在530 eV处观察到新的氧(O 1s)峰.当凝固浴由水换成乙醇后,M2膜表面在397和100 eV处分别观察到属于氮(N 1s)和硅(Si 2p)的两个新峰.此外,M2膜氧(O 1s)峰的强度较M1膜明显增加,氧原子含量由原来的3.36%增加到13.52%.EDS元素分布图如图8所示,能清楚的在M2膜表面观察到C、N、O、F和Si的均匀固定,N、O和Si的出现源于镶嵌在膜表面的P-PDA微凝胶.EDS与XPS测试结果进一步证明了P-PDA微凝胶成功镶嵌在PVDF膜表面上.
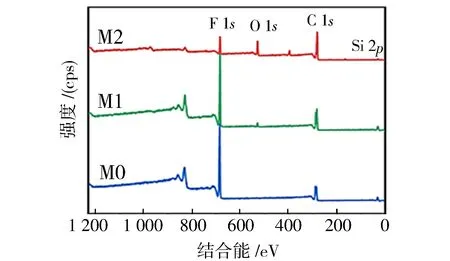
图7 M0膜、M1膜和M2膜的XPS光谱Fig.7 XPS spectra of M0, M1 and M2 membranes

图8 M2膜的元素映射图像(C,N,O,F和Si)Fig.8 Elemental mapping images of M2 membrane (C, N, O, F and Si)
2.4 膜表面浸润性分析
表征了P-PDA微凝胶及乙醇凝固浴对纯PVDF膜及P-PDA微凝胶复合膜表面浸润性的影响.如图9(a)所示,在空气中,M0膜的水接触角为87.2°,由于其含有氟元素,因而表现出一定的疏水性.添加P-PDA微凝胶后,与M0膜相比,M1膜的水接触角略有增加,达到了96.5°[图9(b)],这是因为P-PDA微凝胶通过相转化过程迁移至膜表面,略微增加了膜表面的粗糙度.当凝固浴由水换成乙醇后,由于乙醇凝固浴对膜表面形貌及P-PDA微凝胶迁移过程的巨大影响,导致膜表面粗糙度明显增加,从而显著提高了膜表面的疏水性,M2膜的水接触角达到了147.2°[图9(c)].如图9(d)所示,在空气中,M0膜的油(正己烷)接触角为24.8°.添加P-PDA微凝胶后,M1膜的油接触角略有减小,为19.4°[图9(e)].当凝固浴由水换成乙醇后,M2膜在空气中表现出超亲油性,油接触角为0°[图9(f)].以上结果进一步验证了之前的结论.

图9 (a)M0膜,(b)M1膜和(c)M2膜在空气中的静态水接触角;(d)M0膜,(e)M1膜和(f)M2膜在空气中的油接触角Fig.9 Static water contact angle of (a) M0, (b) M1 and (c) M2 membanes in air; oil contact angle of (d) M0, (e) M1 and (f) M2 membranes in air
为了研究膜的温度/pH响应行为,测量了不同温度和pH条件下的膜表面的浸润性.当温度从25 ℃升高到55 ℃时,M2膜表面的瞬时水接触角都在增加,在30~35 ℃的范围内观察到较为明显的变化,表明在该区域存在LCST转变,这与PNIPAM的相变温度约为32 ℃相一致[图10(a)].此外,由于PMAA中羧基基团的去质子化,羧基变成羧酸根阴离子,导致电荷的形成,从而改变M2膜表面的电荷.膜表面电荷的改变会导致膜表面能增加,从而使M2膜的瞬时水接触角随着pH的增大而减小[图10(b)][35].为了进一步研究水与膜表面之间的动态相互作用,在55 ℃下使水滴(3 μL)与膜表面接触.水滴在离开M0膜时由于水与膜之间的相互作用相对较强留在了膜表面[图10(c)].然而,当离开M2膜表面时,水滴可以很容易地随注射器一起被拉起,表明其对水的亲和力较低,超疏水性能优异.进一步测试了M2膜的pH响应性,如图10(d)所示,当pH=7的水滴在25 ℃时与空气中的膜表面接触时,静置600 s后保持球形,表现出高疏水性(144.3°).相反,pH=7的水滴在空气中14 s内完全扩散并渗透至膜中,使M2膜比中性条件下更亲水.此外,用pH=13的水(浸泡10 min)处理的M2膜表现出优异的水下超疏油性.水下轻油(正己烷)和重油(二氯甲烷)的油接触角(OCA)分别可达156.3°和153.5°[图10(e)和10(f)].
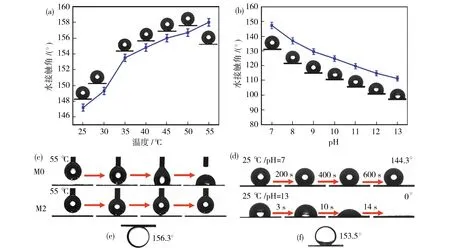
图10 (a)M2膜表面在不同温度下的瞬时水接触角,(b)M2膜表面在不同pH下的瞬时水接触角,(c)M0膜和M2膜在55 ℃下与水滴的动态接触过程,(d)在25 ℃时,pH=7和pH=13的水滴分别与M2膜表面接触后接触角随时间的变化图,(e)用pH=13的水预处理的M2膜的水下轻油(正己烷)接触角,(f)用pH=13的水预处理的M2膜的水下重油(二氯甲烷)接触角Fig.10 (a) Instantaneous water contact angle of M2 membrane surface at different temperatures, (b) instantaneous water contact angle of M2 membrane surface at different pH, (c) dynamic contact process of M0 and M2 membranes with water droplets at 55 ℃, (d) the change of contact angle with time after the water droplets of pH=7 and pH=13 come into contact with M2 membrane surface at 25 ℃, (e) the under-water light oil (n-hexane) contact angle of M2 membrane pretreated with water at pH=13, (f) the under-water heavy oil (dichloromethane) contact angle of M2 membrane pretreatedwith water at pH=13
2.5 膜的自清洁性能
在实际应用中,膜污染会降低分离性能,缩短膜的使用寿命.由于膜表面在pH/温度刺激下具有优异的可切换浸润性,M2膜在自清洁领域的应用前景广阔.为了证明这一点,将附有一滴泵油的膜浸入水中以测试其脱油性能.如图11(a)所示,油滴牢固的粘在M0膜表面,没有丝毫从膜表面脱附的趋势.对于M1膜[图11(b)]和未用pH=13的水预处理的M2膜[图11(c)],油滴没有脱落,依旧是坚固的粘在膜表面.然而,对于用pH=13的水预处理的M2膜,由于其具有优异的水下超疏油性,当被油污染的表面接近水时,表面与水分子接触的地方会发生强烈的水合作用[36].结果,油分子开始从油层边缘解吸并逐渐向中心延伸,然后形成球形液滴并最终脱离膜表面[图11(d)].这些结果表明,M2膜具有优异的自清洁性能,有望有效解决膜在实际应用中的膜污染问题.

(a)M0,(b)M1,(c)未用pH=13的水预处理的M2,(d)用pH=13的水预处理的M2图11 不同膜在中性水中浸泡时的脱油过程图Fig.11 Process diagram of oil removal performance of different membranes soaked in neutral water
2.6 膜的孔径分布
在油水分离过程中,表面结构(孔径和孔隙率)对分离性能起着至关重要的作用.孔径大小和孔隙率对单相(水或油)透过膜的速率有很大影响.如图12(a)所示,M0膜大部分孔径分布在230 nm左右,最大孔径不大于250 nm,最小孔径不小于100 nm.添加P-PDA微凝胶后,与M0膜相比,M1膜孔径略有增大,大部分孔径分布在270 nm左右,最大孔径不大于300 nm,最小孔径不小于100 nm[图12(b)].当凝固浴由水换成乙醇后,由于乙醇凝固浴对NIPS过程的显著影响,M2膜生成大孔结构,大部分孔径分布在1.91 μm左右,最大孔径不大于2.20 μm,最小孔径不小于500 nm[图12(c)].

图12 (a)M0膜、(b)M1膜和(c)M2膜的孔径分布Fig.12 The pore size distribution of (a) M0, (b) M1 and (c) M2 membanes
2.7 膜的油水分离性能测试
到目前为止,大多数已报道的特殊浸润性油水分离膜只具备单一的油水乳液分离性能.在这方面,可实现对不同类型的油水乳液进行按需选择性分离的刺激响应膜在实际复杂的油/水处理中具有很大的潜力.在空气中,M2膜具有高疏水性(147.2°),当膜被pH=13水预处理后,空气中的高疏水性转变为亲水性和水下超疏油性.由于其具有可切换的浸润性,仅在重力驱动下成功实现了对W/O和O/W乳液的有效分离.图13(a)和13(b)展示了在重力驱动下,M2膜对于甲苯包水和水包甲苯乳液的分离过程图.如图13(a)所示,透明的甲苯可以容易地透过M2膜,而水被阻挡.至于水包甲苯乳液的分离,用pH=13水预处理的膜仅允许水通过,而甲苯被截留[图13(b)].为了测试分离效果,拍摄了分离前后进料液和滤液的光学显微图像及实物图.如图13(c)和13(d)所示,分离前的乳液呈现出乳白色,分离后的滤液则是澄清透明的.从光学显微镜中也可以观察到,分离前的甲苯包水和水包甲苯乳液中存在着明显的大液滴,分离后的滤液中则并未观察到明显液滴的存在.上述特性赋予了M2膜在实际按需选择性分离中的潜在用途.

图13 (a)甲苯包水乳液和(b)水包甲苯乳液分离过程的照片,(c)甲苯包水乳液和(d)水包甲苯乳液分离前后的光学显微镜图像及实物图Fig.13 Photographs of (a) water-in-toluene emulsion and (b) toluene-in-water emulsion separation process, (c) optical microscope images and physical images of water-in-toluene emulsion and (d) toluene-in-water emulsion before and after separation
此外,为了进一步评估M2膜的油水分离性能,制备了不同表面活性剂稳定的W/O和O/W和乳液(油包括正己烷、石油醚、液体石蜡和甲苯).表1列出了上述油的物理特性(密度和黏度).如图14(a)所示,M2膜对(正己烷、石油醚、液体石蜡和甲苯)包水的乳液的渗透率分别为458.6、483.3、388.8和443.69 L/(m2·h),分离效率均大于98.5%.我们不难发现,对于较低黏度的油,M2膜的渗透率较高.黏度在油包水乳液的渗透率中起着重要作用:低黏度的油具有小的流动阻力[37-38].当油的黏度增加时,渗透阻力增加,渗透率从大到小依次为:石油醚、正己烷、甲苯和液体石蜡.如图14(b)所示,对水包(正己烷、石油醚、液体石蜡和甲苯)的乳液的渗透率分别为493.6、518.9、482.8和467.7 L/(m2·h),分离效率均在99%左右.在分离O/W乳液的过程中,油的密度越大,越容易沉积在膜表面形成含油滤饼,阻挡水的渗透,从而导致膜的渗透率下降[39-40].结果表明,M2膜的渗透率从大到小依次为石油醚、正己烷、液体石蜡和甲苯.

表1 实验中使用的油的密度和黏度

图14 M2膜对不同类型的(a)油包水乳液和(b)水包油乳液的渗透率及分离效率Fig.14 The permeability and separation efficiency of M2 membrane for different types of (a) water in oil emulsions and (b) oil in water emulsions
3 结论
本研究采用乙醇作为凝固浴通过相转化的方式制备出了一种具有pH和温度双刺激响应性的微凝胶复合膜.在以PVDF为基膜材料的铸膜液的相分离过程中,由于乙醇的弱凝结作用及其对微凝胶具有更好的亲和力,在延缓膜相转化所需时间的同时改善了微凝胶对凝固浴的亲和力,促使更多的微凝胶迁移至膜表面,并为膜表面自由能和膜微纳粗糙结构的同步调控及刺激响应开关的构建提供了保障.制备的膜在空气中表现出高疏水性(147.2°)和超亲油性(0°).在pH=7时,随着温度的升高,膜的水接触角不断增加,并且在30~35 ℃区间内观察到明显的变化,水接触角从149.3°上升到153.5°.当膜被pH=13水预处理后,空气中的高疏水性和超亲油性转变为亲水性和水下超疏油性,水下轻油(正己烷)和重油(二氯甲烷)的OCA分别可达156.3°和153.5°.由于其具有可切换的浸润性,仅在重力驱动下成功实现了对W/O和O/W乳液的有效分离,分离效率均大于98.5%.此外,该膜还具有优异的自清洁性能,有望有效解决膜在实际应用中的膜污染问题.
猜你喜欢
云南化工(2021年11期)2022-01-12 06:06:42
中华养生保健(2020年9期)2021-01-18 03:12:36
无机化学学报(2019年2期)2019-02-27 06:53:38
中国医学影像学杂志(2018年9期)2018-10-17 01:26:52
作文周刊·小学一年级版(2016年23期)2017-06-05 23:27:03
中南大学学报(自然科学版)(2016年2期)2017-01-19 07:37:19
癌症进展(2016年8期)2016-08-22 11:22:06
山东医药(2015年14期)2016-01-12 00:39:54
质量与标准化(2015年12期)2015-07-10 15:11:50
应用化工(2014年7期)2014-08-09 09:20:27
