
α-二亚胺镍(Ⅱ)催化乙烯与乙烯基三甲氧基硅烷共聚
2023-11-06 12:34:24张丹枫张文杰
华东理工大学学报(自然科学版) 2023年5期
张丹枫, 张文杰, 牛 犇, 仝 鑫
(华东理工大学材料科学与工程学院, 上海市先进聚合物材料重点实验室, 上海 200237)
聚烯烃的非极性极大地限制了其最终应用,而在聚烯烃链中引入少量功能性基团就能够使聚合物的性能及商业价值得到很大提升,因此如何高效低廉地将功能性基团引入到聚烯烃链中得到功能化聚烯烃成为了当前研究热点之一[1-4]。近年来,随着后过渡金属催化剂研究的突破,其较弱的亲氧性,使得其催化乙烯与极性单体共聚制备功能性的聚烯烃材料逐渐成为可能。其中常见的功能性基团主要有羧基[5]、羟基[6-8]、酯基[9-11]、氰基[12]、卤素[13-14]和硅[15-19]等。由于硅原子对催化剂的毒害作用较小,且有机硅聚合物具有独特的理化性质,因此含硅功能化聚烯烃在聚烯烃改性领域占有重要地位[20-21]。
2004 年,Ciolino 等[22]采用EtInd2ZrCl2/MAO 催化体系在90 ℃和110 ℃下催化乙烯/乙烯基聚(二甲基硅氧烷)(PDMS)共聚得到了分子链中含有Si-O-Si 的共聚物。硅单体的加入会明显降低催化体系的催化活性,在聚合温度为110 ℃时尤为明显。90 ℃聚合温度下所得共聚物的共聚单体含量、共聚物分子量、催化活性均高于110 ℃下所得共聚物。随后,Ciolino 等[23]在sec-BuLi 的存在下使用两步法催化丁二烯和六甲基环三硅氧烷共聚得到了结构均匀和分子量分布较窄的聚 (1,4-丁二烯) (PB) 和PDMS 的二嵌段共聚物PB-b-PDMS。所得共聚物的热稳定性随着PDMS 含量的增加而增加。透射电子显微镜结果表明,共聚物具有不同的形态,从球形到层状不等,甚至是圆柱形形态。Zimmer 等[24]合成了一种新型乙烯基硅氧烷单体,二叔丁氧基(甲基)(辛-7-烯基)硅烷,并采用rac-EtInd2ZrCl2/MAO催化体系催化其与丙烯共聚。结果表明,共聚物的链长越长,聚合物链中的硅烷单体插入率越高。所得共聚物经过三氟甲磺酸处理后可以使叔丁氧基硅烷官能团发生不同程度的交联,进而获得不溶性聚合物材料。这种方法可以控制共聚物组成,从而控制共聚物的交联程度。2014 年,Xu 等[25]利用乙烯基封端聚乙烯(PE)的硫醇-烯点击化学和酯化反应制备了PE-b-PDMS 共聚物。共聚物中PDMS 含量越高,PE 链段的结晶度和规整度越低,共聚物的热稳定性随着PDMS 含量的增加而增加。这种二嵌段共聚物可用作高密度聚乙烯和硅油共混物的增溶剂:当引入PEb-PDMS 作为增溶剂时,共混物的断裂伸长率明显增加,这表明即使加入较少的PE-b-PDMS,该二嵌段共聚物也能够桥接基质和分散相。
后过渡金属催化剂由于对杂原子的耐受能力较强,因此常用于催化乙烯和功能性单体共聚[26-28]。2017年,Chen 等[29]使用两种 (α-二亚胺)Ni(Me)(CH3CN)+/B(C6F5)3催化体系催化乙烯和乙烯基三乙氧基硅烷(VTEoS)共聚,得到了高分子量共聚物。调节催化剂的种类和反应条件,可以得到微观结构各异的共聚物(从主链接近线性到高度支化)。每条聚合物链含有多个-Si(OEt)3基团,共聚单体VTEoS 的插入率可达到10.0%(摩尔分数)。此外他们还对聚合机理进行了研究,低温核磁共振结果表明,VTEoS 以2,1-和1,2-方式插入Ni-R 键,得到四元和五元螯合物,这些螯合物与乙烯快速反应生成“开环”乙烯烷基络合物。通过调整乙烯压力和温度可以调整-Si(OR)3基团在聚合物链内和聚合物链端的比例,该比例随着乙烯压力的增加而增加,随着温度的增加而减少。
近年来,本实验室主要致力于过渡金属催化剂的设计与合成,及其催化烯烃聚合、烯烃与功能性单体共聚方面的研究,包括水杨醛亚胺镍系配合物催化乙烯聚合[30]、MMA 聚合[31]、乙烯与MMA 共聚[9,32-33],α-二亚胺镍催化剂催化乙烯聚合[34-35]、MMA 聚合[36],环戊二烯基镍催化剂催化苯乙烯[37]、乙烯[38]聚合,以及“一锅法”N-(2-苯甲酰胺苯基)-水杨醛亚胺-TiCl4·2THF 催化乙烯聚合[39]等。本文以α-二亚胺镍(Ⅱ)催化剂催化乙烯和乙烯基三甲氧基硅烷(VTMoS)共聚,研究了不同聚合条件和不同催化剂结构对共聚反应在催化活性和共聚物微观结构等方面的影响。
1 实验部分
1.1 原料和试剂
2,6-二异丙基苯胺、蒽醌、乙二醇二甲醚、六水合溴化镍、二苯甲酮、无水硫酸镁、无水乙醚、二氯甲烷、甲酸和无水乙醇均为分析纯,上海凌峰化学试剂有限公司,其中乙二醇二甲醚和无水乙醚在氩气保护下经钠丝-二苯甲酮回流至紫色,蒸出备用;二氯甲烷在氩气氛围下使用氢化钙回流数小时,蒸出备用。乙二醛水溶液(w=40%)、氯化锌、甲苯和对二甲苯均为分析纯,国药集团化学试剂有限公司,其中甲苯在氩气氛围下经钠丝-二苯甲酮回流至紫色,蒸出备用。2,3-丁二酮、2,6-二甲基苯胺,纯度99%,上海泰坦科技股份有限公司,直接使用。冰醋酸,分析纯,上海天莲化工科技有限公司,直接使用。倍半乙基氯化铝、w=10%甲苯溶液,南京通联化工有限公司,直接使用。乙烯基三甲氧基硅烷,纯度99.9%,上海毕得医药科技有限公司,直接使用。乙烯,纯度99%,上海春雨特种气体有限公司,聚合前经4A 分子筛纯化后使用。
1.2 测试与表征
Bruker Avance-400MHz 核磁共振波谱仪(美国Bruker 公司),以1, 2, 4-三氯苯和氘代邻-二氯苯(体积比为1∶1)为溶剂,在125 ℃下测定;Nicolet 5700 红外光谱仪(Thermo Electron 公司),溴化钾压片;MDSC TA-2900 差示扫描量热仪(美国TA 仪器公司),N2气氛,升温及降温速率均为10 ℃/min;等离子体发射光谱仪(ICP,波长范围167~785 nm,725 型,美国Agilent 公司)。
1.3 实验步骤
1.3.1 催化剂的合成 参考文献[40]方法制备α-二亚胺配体及其对应的镍催化剂[(2,6-R2)-C6H3-N=C(R1)-C(R1)= N-C6H3-(2,6-R2)]NiBr2(C1: R1= H,R2= CH3;C2: R1= H, R2= CH(CH3)2;C3: R1= CH3,R2= CH3; C4: R1= CH3, R2= CH(CH3)2; C5: R1=acenaphthyl, R2= CH(CH3)2)(图1),采用核磁(1HNMR)与元素分析方法分别表征其结构。
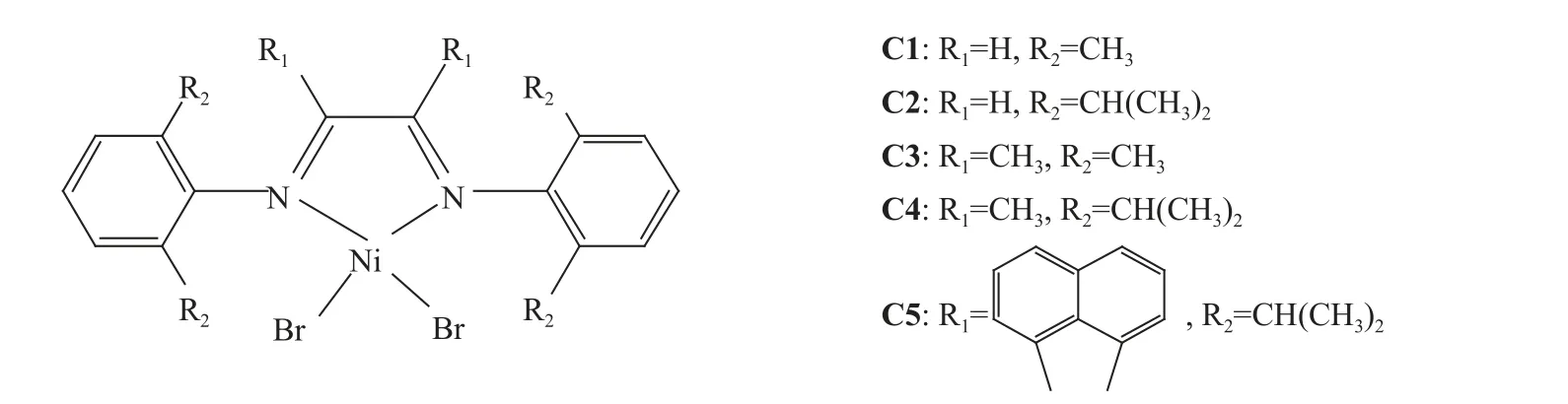
图1 α-二亚胺镍化合物Fig.1 α-Diimine nickel(II) complexes
1.3.2 乙烯/VTMoS 共聚 在150 mL 清洁干燥的不锈钢高压釜中进行乙烯和VTMoS 的共聚反应,采用油浴控温。聚合前先抽烤反应釜,冷却后用氩气和乙烯置换,并在乙烯保护下依次加入催化剂、溶剂和VTMoS,通乙烯至饱和后加入助催化剂倍半乙基氯化铝(EASC),调节乙烯至所需压力,继续通入乙烯,反应一定时间。用乙醇溶液终止反应,将所得聚合物过滤,用无水乙醇洗涤,真空干燥至恒重。
1.3.3 乙烯/VTMoS 共聚物凝胶含量的测定 由于所得共聚物的弹性较好、溶解性较差,不能采用常规的高温凝胶渗透色谱(GPC)、乌氏黏度计等方法测定共聚物的分子量,因此对乙烯/VTMoS 共聚物的溶剂吸收系数(f)、凝胶质量分数(wgel)以及有效链平均分子量(Mc)进行分析与表征[41]。具体步骤为:将共聚物(共聚物溶胀前起始质量m0为10~20 mg)加入到对二甲苯(V=50 mL)中,139 ℃下回流5 h,趁热过滤,称量得到共聚物溶胀后的质量msw,真空干燥直至恒重得到质量mgel。利用式(1)~式(5)计算溶剂吸收系数、凝胶含量和有效链平均分子量。
其中:φpol为凝胶中聚合物的体积分数;V0为溶剂的摩尔体积,V0= 139.3 cm3/mol;ρpol为聚合物密度,ρpol=0.806 g/cm3(139 ℃);ρsolv为溶剂密度,ρsolv= 0.761 g/cm3(139 ℃);c为有效浓度;μ为Huggins 溶剂/聚合物相互作用参数,μ=0.31。f越大,意味着Mc越大,交联强度越小;wgel越大,共聚物的交联网络越均匀。
2 结果与讨论
2.1 乙烯/VTMoS 共聚的最优化催化工艺条件
首先,以催化剂C4 为主催化剂,EASC 为助催化剂,考察了VTMoS 浓度、催化剂物质的量、nAl/nNi、乙烯压力、聚合时间和溶剂等变化因素对乙烯/VTMoS 共聚的影响,结果见表1,以催化活性为主要考察目标,筛选出乙烯/VTMoS 共聚反应的最优化催化工艺条件。
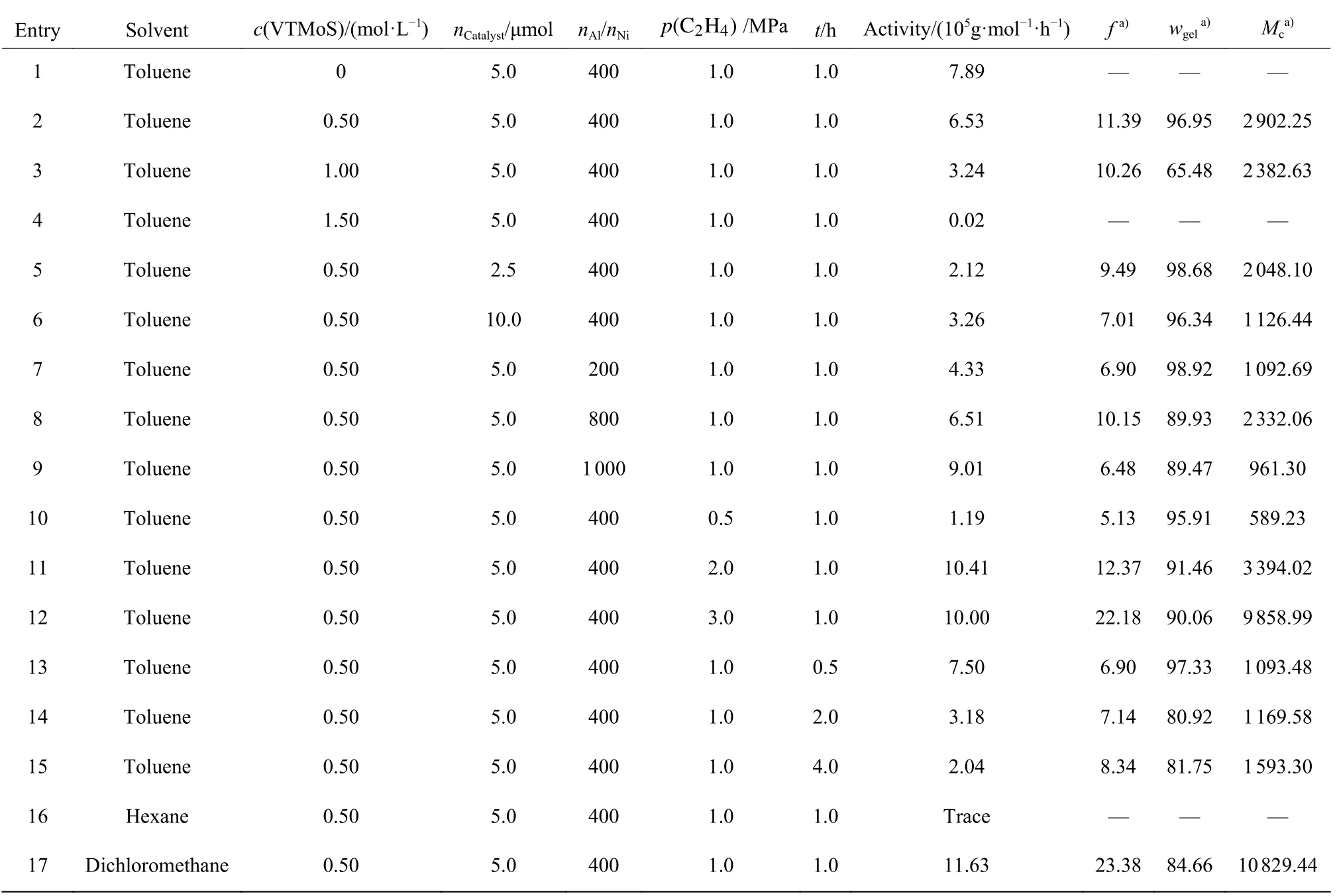
表1 反应条件对催化剂C4 催化乙烯/VTMoS 共聚的影响Table 1 Influence of reaction conditions on copolymerization of ethylene/VTMoS by catalyst C4
由表1 中Entry 1 ~ 4 可知,VTMoS 浓度对乙烯/VTMoS 共聚有显著影响。随着VTMoS 浓度增加,催化活性逐渐降低。当无VTMoS 时,催化活性为7.89×105g/(mol·h);当VTMoS 浓度为0.50 mol/L时,催化活性降至6.53×105g/(mol·h);当VTMoS浓度增加至1.00 mol/L 时,催化活性降至3.24×105g/(mol·h);当VTMoS 浓度进一步增加至1.50 mol/L时,催化活性降至最低,为0.02×105g/(mol·h)。这表明VTMoS对催化剂活性中心有一定的毒害作用。另外,VTMoS 浓度对共聚物的溶剂吸收系数、凝胶含量和有效链平均分子量也有较大的影响。随着VTMoS浓度的增加,溶剂吸收系数和有效链平均分子量下降,表明共聚物交联程度增加。因此,最优化的VTMoS 浓度为0.50 mol/L。
由表1 中Entry 2、Entry 5 和Entry 6 可知,随着催化剂物质的量的增加,催化活性具有先增加后下降的趋势。当催化剂物质的量从2.5 μmol 增加至5.0 μmol 时,催化剂的活性从2.12 × 105g/(mol·h)增加至6.53 × 105g/(mol·h)。这是因为随着催化剂物质的量的增加,体系中催化活性中心数量增加,活性也相应增加。进一步增加催化剂的物质的量至10.0 μmol时,催化体系可能因反应速度加快而使体系交联程度增加,从而阻止了体系中乙烯和VTMoS 单体向活性中心的扩散并插入,导致催化活性下降至3.26 ×105g/(mol·h)。因此,催化剂最优物质的量为5.0 μmol。相应地,随着催化剂物质的量的增加,所得共聚物的溶剂吸收系数、凝胶含量均表现出先增加后下降的变化趋势,而有效链平均分子量则逐渐下降。当催化剂的物质的量增加至10.0 μmol 时,溶剂吸收系数降至7.01,Mc降至1 126.44。在催化剂用量为2.5~10.0 μmol/L 时凝胶质量分数均比较高,达到96.34% ~98.68%,表明交联程度比较均匀。
对比表1 中Entry 2、Entry 7~Entry 9 可知,nAl/nNi对催化活性和共聚物微观结构有较大的影响。催化活性总体上随着nAl/nNi增加而增加。当nAl/nNi=200 时,催化活性为4.33 × 105g/(mol·h);nAl/nNi为400、800 时,催化活性分别为6.53 × 105g/(mol·h)和6.51 × 105g/(mol·h);而当nAl/nNi= 1 000 时,催化活性达到9.01 × 105g/(mol·h)。这是因为随着助催化剂EASC 用量增加,体系中活性中心含量也随之增加所致。但所得共聚物的溶剂吸收系数和有效链平均分子量具有先增加后下降的变化趋势。当nAl/nNi=400 时,溶剂吸收系数和有效链平均分子量达到最大,分别为11.39、2 902.25。这表明当助催化剂EASC用量增加时,共聚物发生交联的程度也随之增加,此时凝胶质量分数为89.47% ~ 98.92%,共聚物交联网络非常均匀。由此得到最优化的nAl/nNi为 400。
对比表1 中Entry 2、Entry 10~Entry 12 可知,乙烯压力对催化活性和共聚物微观结构也有较大的影响。催化活性随着乙烯压力的增加先增加后略有降低。当压力为2.0 MPa 时,催化活性达到最高,为10.41×105g/(mol·h)。这是因为随着体系中乙烯压力的增加,溶解在溶剂中的乙烯含量增加,有利于聚合反应的进行。而所得共聚物的溶剂吸收系数和有效链平均分子量均随乙烯压力的增加而增加,表明共聚物交联程度降低。当乙烯压力为0.5 MPa 时,溶剂吸收系数仅为5.13,Mc为589.23。但当乙烯压力为3.0 MPa 时,溶剂吸收系数达到22.18,Mc达到9 858.99。此时,凝胶质量分数均比较高,为90.06% ~96.95%,表明交联程度比较均匀。由此得到最优化的乙烯压力为1.0 MPa。
由表1 中Entry 2、Entry 13~Entry 15 可知,催化活性随聚合时间的延长逐渐下降。当聚合时间为0.5 h 时,催化剂活性最高,为7.50 × 105g/(mol·h);而当聚合时间为4.0 h 时,催化剂活性仅为2.04 ×105g/(mol·h)。所得共聚物的溶剂吸收系数和有效链平均分子量规律不明显。当聚合时间从0.5 h 延长至4.0 h 时,溶剂吸收系数在6.90 ~ 11.39 之间,而有效链平均分子量为1 093.48 ~ 2 902.25。其中聚合时间为1 h 时,溶剂吸收系数和有效链平均分子量达到最大,分别为11.39 和2 902.25。这表明聚合时间的延长导致共聚物发生交联程度增加。而凝胶质量分数则随着聚合时间的延长总体上呈下降趋势,变化范围为80.92% ~ 97.33%,表明共聚物交联网络随聚合时间的延长逐渐趋于不均匀。由此得到最优化的聚合时间为1.0 h。
由表1 中Entry 2、Entry 16 和Entry 17 可知,反应溶剂对催化活性具有较大影响。与相同条件下以甲苯为溶剂的聚合结果相比,以二氯甲烷作为溶剂时具有更高的催化活性(11.63 × 105g/(mol·h)),以及更高的溶剂吸收系数(23.38)和有效链平均分子量(10 829.44),即更低交联程度以及较均匀的交联网络;但以正己烷为溶剂时则聚合效果不佳,仅得到微量的聚合物。因工业化应用对于溶剂的安全性和毒性等要求,故最佳的聚合溶剂为甲苯。
综上,以C4 为催化剂、EASC 为助催化剂进行乙烯/VTMoS 共聚反应的最优化催化工艺条件为:VTMoS 浓度为0.50 mol/L,催化剂物质的量为5.0 μmol,nAl/nNi= 400,乙烯压力为1.0 MPa,聚合时间为1.0 h。基于最优化催化工艺条件,进一步考察了甲苯溶剂中催化剂结构对共聚反应的影响,结果见表2。
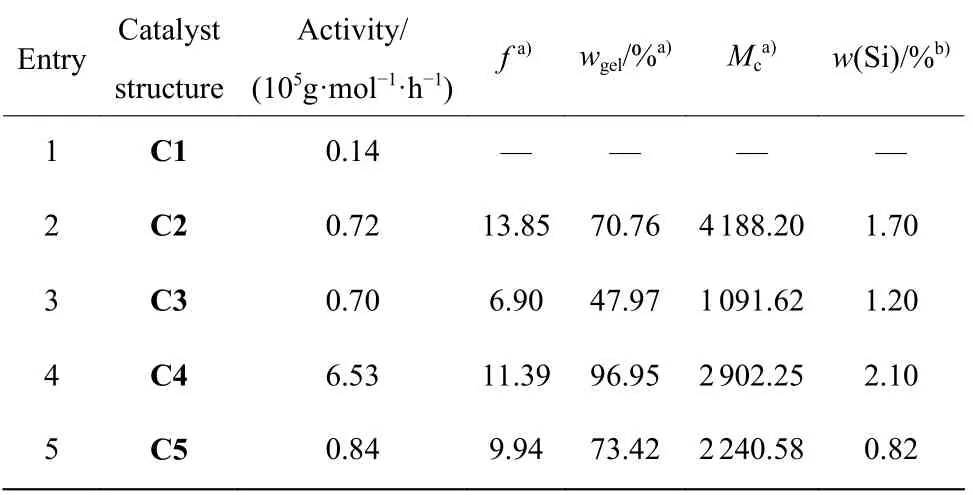
表2 催化剂结构对乙烯/VTMoS 共聚的影响Table 2 Influence of catalyst structures on ethylene/VTMoS copolymerization
由表2 可知,不同催化剂结构对催化活性和共聚物的微观结构具有显著影响。首先,在甲苯溶剂中,C4 的催化活性最高,为6.53 × 105g/(mol·h),是催化剂C2、C3 和C5 催化活性的10 倍左右。这可能与催化剂在甲苯中的溶解性有关,其中催化剂C4 的溶解性最好,催化剂C1 的溶解性最差。其次,从共聚物的微观结构看,随着α-二亚胺骨架结构位阻的增加,共聚物溶剂吸收系数和有效链平均分子量随之降低。如Entry 2、Entry 4 和Entry 5 所对应的催化剂C2、C4 和C5 所得的共聚物溶剂吸收系数分别为13.85、11.39 和9.94,有效链平均分子量分别为4 188.20、2 902.25、2 240.58。这表明α-二亚胺骨架结构的增加将增加共聚物的交联程度。此外,从所得共聚物的硅含量(质量分数,下同)看,其中以C4 的共聚物硅含量最高,达2.10%,其次为C2(1.70%),再次为C5(0.82%)。这表明随着α-二亚胺骨架结构位阻的增加,共聚物硅含量增加,但位阻进一步增加,可使共聚物硅含量降低。因此,最优化的催化剂结构为C4。
2.2 乙烯/VTMoS 共聚物结构
对所得乙烯/VTMoS 共聚物的结构分别采用高温1H-NMR 和13C-NMR、傅里叶红外光谱(FT-IR)进行了分析与表征,结果见图2~图4。

图2 乙烯/VTMoS 共聚物的1H-NMR 谱图(表2 中Entry 4)Fig.2 1H-NMR of ethylene/VTMoS copolymer (Entry 4 in table 2)
图2 和图3 分别是表2 中Entry 4 所得共聚物的1H-NMR 和13C-NMR。由图2 可知,化学位移0.97 ~1.39 处是共聚物中甲基、次甲基、亚甲基上氢的特征峰,化学位移3.48 ~ 3.93 处是共聚物中-Si(OCH3)3上氢的特征峰,化学位移7.07 ~ 7.36 处是溶剂峰。由图3 的13C-NMR 可知,化学位移11.42 ~ 39.91 处是共聚物中聚乙烯链上碳的特征峰,呈支化聚乙烯的结构,化学位移128.18 ~ 134.17 处是溶剂峰。结合共聚物的1H-NMR、13C-NMR 谱图以及表1、表2 中的溶剂吸收系数可知,所得共聚物为含硅、含支链的乙烯/VTMoS 共聚物。
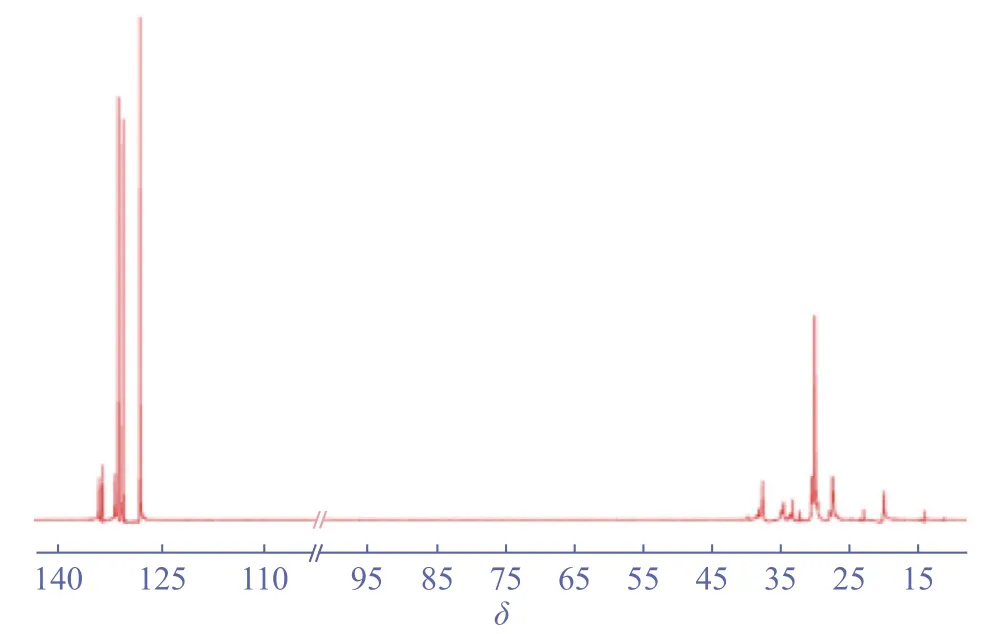
图3 乙烯/VTMoS 共聚物的13C-NMR 谱图(表2 中Entry 4)Fig.3 13C-NMR of ethylene/VTMoS copolymer (Entry 4 in table 2)
图4 是表2 中Entry 5 的乙烯/VTMoS 共聚物的红外谱图。3 426 cm-1处为-OH 基(包括Si-OH 基)的伸缩振动峰,2 919 cm-1处为-CH2-的不对称伸缩振动峰,2 850 cm-1处为-CH2-的对称伸缩振动峰,1 471 cm-1处为-CH2-的剪式变角振动峰,1 092 cm-1处为Si-O-C、Si-O-Si 的振动峰,801 cm-1处为Si-OH 的振动峰,719 cm-1处为碳原子数大于4 的-CH2-的平面摇摆振动峰,表明该聚合物为具有一定交联程度的乙烯/VTMoS 共聚物。
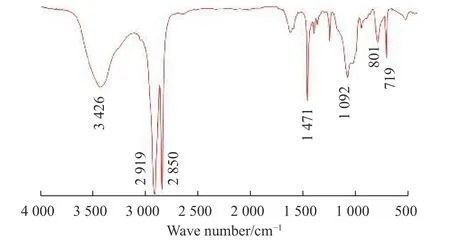
图4 乙烯/VTMoS 共聚物的红外谱图(表2 中Entry 5)Fig.4 FT-IR spectrum of ethylene/VTMoS copolymer (Entry 5 in table 2)
2.3 乙烯/VTMoS 共聚物热性能
对所得乙烯/VTMoS 共聚物的热性能采用差示扫描量热仪进行了测试。图5 示出了C3~C5 所得乙烯/VTMoS 共聚物的DSC 图。C3 和C5 所得共聚物的熔点分别为107.04 ℃和109.28 ℃,且熔融峰较窄,而C4 所得共聚物的熔点不明显(46.99 ℃),熔融峰较宽。这可能与3 种共聚物相应的硅含量、共聚物交联程度及共聚物交联网络的均匀性密切相关,其中C4 所得共聚物的硅含量较高,共聚物交联程度较高且共聚物交联网络更均匀。

图5 乙烯/VTMoS 共聚物的DSC 图(表2 中Entry 3~5)Fig.5 DSC curves of ethylene/VTMoS copolymers (Entry 3~5 in table 2)
3 结 论
以催化剂C4 为模型催化剂,EASC 为助催化剂,考察了VTMoS 浓度、催化剂物质的量、nAl/nNi、乙烯压力、聚合时间和溶剂等变化对乙烯/VTMoS 共聚的影响,筛选得到乙烯/VTMoS 共聚反应的最优化催化工艺条件为:VTMoS浓度为0.50 mol/L,催化剂物质的量为5.0 μmol,nAl/nNi为400,乙烯压力为1.0 MPa,聚合时间为1.0 h。在聚合温度为25 ℃和最优化催化工艺条件下,C4 催化剂的催化活性达到6.53 ×105g/(mol·h),共聚物中硅质量分数为 2.10%。随着α-二亚胺骨架结构位阻的增加,共聚物的交联程度增加,交联网络更加均匀。
猜你喜欢
纺织科学研究(2021年7期)2021-08-14 01:42:30
原子与分子物理学报(2021年2期)2021-03-29 07:31:46
现代检验医学杂志(2016年1期)2016-11-12 13:19:54
国外医药(抗生素分册)(2016年4期)2016-07-12 14:25:21
当代化工研究(2016年2期)2016-03-20 16:21:20
中国塑料(2015年6期)2015-11-13 03:02:55
化工进展(2015年3期)2015-11-11 09:18:44
材料研究与应用(2015年4期)2015-08-23 11:39:36
中国当代医药(2015年7期)2015-03-01 02:01:21
应用化工(2014年9期)2014-08-10 14:05:08
