
超弹性自修复导电水凝胶的制备及性能
2023-11-06 12:34:20赵博文
华东理工大学学报(自然科学版) 2023年5期
赵博文, 张 静, 张 琰
(1.华东理工大学材料科学与工程学院, 上海 200237;2.上海市纳米科技与产业发展促进中心, 上海 200237)
导电水凝胶具有刺激响应电传感特性,可以将应变[1]、浓度[2]、温度[3]、pH 值[4]等外界刺激转化成可检测的电信号[5-7],因此,被广泛应用于可穿戴传感设备中,用于运动记录和健康监测。然而传统的导电水凝胶化学交联网络不均匀,使得导电水凝胶在外力加载过程中耗散能量的能力变差,结构脆弱并且容易破碎[8],导致其在低应变下就开始产生断裂,显现出较低的抗拉强度和韧性。
近几年,基于相互渗透的双网络水凝胶和半互穿网络水凝胶概念变得十分流行。双网络水凝胶通过将两个互相渗透的网络组合在一起,形成了一种具有极高拉伸强度和韧性的水凝胶[9]。半互穿网络水凝胶通过配位键[10]、氢键[11]、主客体相互作用[12]以及聚合物链之间形成的离子相互作用[13]等一系列的可逆非共价键相互作用,获得了优秀的能量耗散能力,并具有了自修复能力[14]。水凝胶的自修复机制除了上述所说的非共价键外,还存在例如席夫碱[15]、硼酸酯键[16]和Diels-Alder(环加成反应)[17]等动态共价键的修复机制。但是单单引入一种自修复机制的水凝胶,往往存在很多缺陷,如:基于席夫碱反应的自修复水凝胶通常需要将样品加热到70 ℃以上,然后再冷却以实现可逆的结合和自修复[18-20];基于氢键的自修复材料具有较强自修复效率,但由于氢键相互作用力较弱,导致以氢键为主体的自修复水凝胶的机械强度较弱[21-24]。非共价键的自修复水凝胶通常较软且机械强度相对较低,而以单一动态共价键为基础的自修复水凝胶会缺乏弹性,其修复条件苛刻且修复效率低[18]。通过结合动态共价键和非共价键的作用力,可以在提高力学性能的同时获得满意的修复效果。Dai 课题组[25]通过简单的交联反应,依靠动态硼酸酯键和氢键两种动态愈合机制的形成,制备了力学性能和自修复性能良好的聚乙烯亚胶-聚乙烯醇-甲酰基苯基硼酸(PEI-PVA-Bn)水凝胶。Zhang 课题组[26]通过动态硼酸酯键和氢键的协同作用,制备了具有较高机械强度、较高韧性和抗疲劳性能的水凝胶,在室温下具有良好的自修复性能。
为了解决目前由单一自修复机制制备的自修复导电水凝胶修复效率差、机械强度低的问题,本文采用一步法基于丙烯酰胺(AM),N-2-氨基乙基丙烯酰胺盐酸盐(AEAM)和N-丙烯酰氧基琥珀酰亚胺(ASI)3 种单体制备了P(AM-AEAM-ASI)/BORAX(硼砂)-LiCl(下文简称PAESB-L)导电水凝胶。ASI 作为一种单体,常用于多肽或蛋白质中与氨基形成席夫碱[27];AEAM 作为一种聚电解质,提供氨基与ASI 反应生成席夫碱,同时为水凝胶提供导电性能[28]。水凝胶通过席夫碱和氢键的多重相互作用自修复,不仅降低了水凝胶的自修复条件,还增加了水凝胶的柔韧性。此外,聚电解质AEAM 和电解质LiCl 增强了水凝胶的导电性能。
1 实验部分
1.1 原料和试剂
二氯甲烷(DCM),分析纯,纯度≥99%;甲醇,分析纯,纯度≥99%;异丙醇,分析纯,纯度≥99%,以上均购于国药集团化学试剂有限公司。三乙胺,分析纯,纯度≥99%,上海凌峰化学试剂有限公司。丙烯酰氯,纯度≥99.5%;N-羟基琥珀酯,纯度≥99%;乙二胺,纯度≥99%;乙二胺二盐酸盐,纯度≥99.5%;BORAX,纯度≥99%;LiCl,纯度≥99%,以上均购于Adamas 试剂有限公司。2-羟基-4′-(2-羟乙氧基)-2-甲基苯丙酮(I2959),纯度≥99.5%,购于Sigma Aldrich试剂有限公司。
1.2 水凝胶的制备
1.2.1 AEAM 的制备 AEAM 的合成参考Wu 等[28]的合成方法并做出一些改进,合成路线如图1 所示。

图1 AEAM 单体的合成路线Fig.1 Synthetic routes for monomer AEAM
首先,将乙二胺二盐酸盐(0.226 mol)、去离子水(120 mL)和乙二胺(0.532 mol)混合,加入搅拌子在室温25 ℃下搅拌1 h,加入160 mL 甲醇后,再继续搅拌1 h,然后将反应体系置于-30 ℃的低温反应槽中,待反应体系稳定在-30 ℃后缓慢滴加丙烯酰氯(0.532 mol)。在丙烯酰氯完全加入后,将反应体系升温至-20 ℃继续反应1 h,然后加入质量分数37%的浓盐酸,调节溶液的pH 值至1~2。待系统温度升至室温后,使用液氮将溶液冻结,置入冷冻干燥机中除去溶剂。在室温条件下,将反应生成的白色粉末在异丙醇中溶解,然后过滤掉体系内残渣,得到无色透明液体,置于四氢呋喃(THF)中沉降,然后置于真空烘箱中干燥,干燥后的白色粉末再溶于异丙醇中,重复上述沉降步骤两次,最后得到的白色粉末溶于异丙醇,在-20 ℃重结晶,然后置于真空烘箱中干燥至恒重,产率为23.1%。
1.2.2 ASI 的制备 ASI 的制备参考Santos 课题组[29]的合成方法,并进行了一些简化。将N-羟基琥珀酰亚胺(3.3 mmol)和三乙胺(3.3 mmol)溶于无水二氯甲烷(20 mL)中,用冰水浴将混合物冷却到0 ℃,然后滴加丙烯酰氯(3.9 mmol);0.5 h 后,大力搅拌,室温下反应过夜,将产生的白色沉淀过滤,依次用水(50 mL)、饱和碳酸氢钠溶液(50 mL)和水(50 mL)清洗溶液,收集有机相溶液,加入无水硫酸镁除水并进行抽滤,然后将有机溶剂旋蒸除去,得到的淡黄色晶体在40 ℃下溶于乙酸乙酯与正己烷的混合溶液(体积比20∶80)中,待晶体溶解后置于-20 ℃中重结晶,将有机溶剂除去,再将得到的晶体溶于上述浓度的乙酸乙酯与正己烷混合溶液中反复重结晶3 次,最终得到白色的ASI 晶体,产率47.3%。ASI 的合成路线如图2所示。
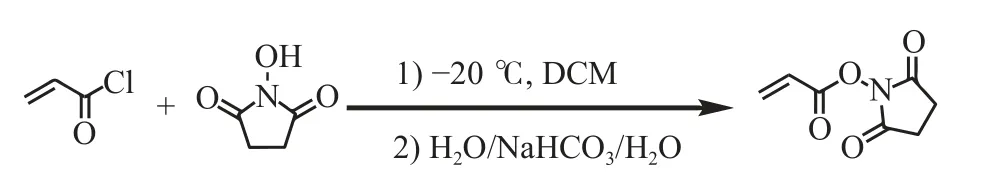
图2 ASI 单体的合成路线Fig.2 Synthetic routes for ASI monomer.
1.2.3 PAESB-L 水凝胶的制备 首先将AM、AEAM、ASI、BORAX 按一定比例溶解到LiCl(2 mol/mL)溶液中,用氩气吹扫20 min,然后加入引发剂I2959 后,用汞灯(紫外光波长284 nm)光刻机EVG620(EVG)固化形成水凝胶。
1.3 测试与表征
1.3.1 AEAM 和ASI 的结构表征 采用核磁共振波谱仪(瑞士Bruker,AV400 型)表征AEAM 和ASI 的结构,外加磁场频率400 MHz。AEAM 的核磁采用D2O 作为溶剂,ASI 的核磁采用氘代氯仿(CDCl3)作为溶剂。
1.3.2 水凝胶的结构表征 水凝胶的结构采用傅里叶红外分析仪(美国尼高力仪器公司 Nicolet 5700 型)进行分析,扫描范围600~4 000 cm-1。先将水凝胶样品冷冻干燥后,将其切片制备成厚约5 mm、直径2 cm的小圆片,通过红外光谱仪上的衰减全反射(ATR)配件测定水凝胶红外光谱,扫描范围600~4 000 cm-1。
1.3.3 水凝胶的拉伸性能测试 在室温条件下,通过20 N 称重传感器的万能拉力机(MTS 系统(中国)公司SANS CMT2503 型)测试水凝胶样品的拉伸性能,将水凝胶两端宽部分别固定在两个聚苯乙烯夹子上,将待测试的凝胶初始长度设置为10 mm,用夹具固定聚苯乙烯夹子,拉伸速度设置为10 mm/min,直到水凝胶样品被拉断为止结束测试。在水凝胶弹性形变范围内应力和应变成正比例关系(即符合胡克定律),其比例系数即弹性模量(E)按照式(1)进行计算,其中σ为应力,ε为应变。
1.3.4 水凝胶的自修复能力测试 采用MTS 系统(中国)公司生产的万能拉力机(SANS CMT2503 型)测试了水凝胶的自修复性能。水凝胶的自修复效率(η)按照式(2)进行计算,其中εt为自修复th 后水凝胶的断裂伸长率,ε0为水凝胶初始的断裂伸长率。
1.3.5 水凝胶导电性能的测试 将水凝胶样品切成长方体块状(12 mm×2 mm×2 mm),与电化学工作站(上海辰华仪器有限公司,Chi760e 型)链接进行检测。链接水凝胶与电化学工作站的电极夹夹住水凝胶两端,间距保持在10 mm。在5 mV 的开路电压下,电化学工作站在10-1~106Hz 的频率范围内进行了电化学阻抗谱图测试,电导率(κ)根据式(3)进行计算,式中L代表两电极夹之间距离,S为水凝胶截面的面积,Rb是内部电阻。
2 结果与讨论
2.1 AEAM 和ASI 的合成
AEAM 经过多次重结晶纯化之后所得到的核磁共振氢谱如图3(a)所示。各质子氢的化学位移如下:氘代试剂D2O 中残余的DOH 的质子氢(斜体)化学位移δ= 4.80,δa,c= 6.15(60 Hz,CH2= CH),δb= 5.70(44 Hz,CH2= CH),δd= 3.50(24 Hz, NHCH2CH2),δe= 3.10(36 Hz, CH2NH2)。此外,上述质子峰的积分面积比Ia,c∶Ib∶Id∶Ie为2∶1∶2∶2,与理论值一致。上述结果表明,AEAM单体结构精确,满足后续反应需求。

图3 AEAM(a)和ASI(b)的核磁共振氢谱Fig.3 1H-NMR spectra of AEAM (a) and ASI (b)
经过反复提纯后的ASI 所得到的核磁共振氢谱如图3(b)所示。各质子氢的化学位移如下:氘代试剂CDCl3中的质子氢(斜体)化学位移δ= 7.27,δa=6.70(36 Hz,CH2= CH),δb= 6.20(36 Hz,CH2= CH),δc= 6.35(44 Hz, CH2=CH),δd= 2.88(68 Hz,CH2CO)。上述质子峰的积分面积比Ia∶Ib∶Ic∶Id为1∶1∶1∶2,与理论值一致。此外,谱图中未出现其他杂质峰。上述结果表明,ASI单体结构精确,满足后续反应需求。
2.2 水凝胶的结构
图4 为聚丙烯酰胺(PAM)、聚(丙烯酰胺-N-丙烯酰氧基琥珀酰亚胺)(PAS)、PAESB-L 水凝胶的红外光谱图。在PAM、PAS、PAESB-L 谱图上2 919 cm-1处均出现了-N-H 的伸缩振动峰,以及1 724 cm-1处C= O 伸缩振动峰和950 cm-1处NCO 弯曲振动峰,证明了丙烯酰胺上酰胺基团的存在。此外,在PAS 上可以看到ASI 上特有的琥珀酰亚胺的叔胺峰,向高场移动出现在1 415 cm-1处。在PAS 和PAESB-L 水凝胶的红外谱图上可以看到在1 301 cm-1处出现-C-O-的伸缩振动峰,PAESB-L 水凝胶在1 041 cm-1处出现-N-O-的伸缩振动峰,证明了席夫碱的形成;PAESB-L 水凝胶上琥珀酰亚胺上特有的叔胺峰消失了,也从侧面证明了席夫碱的形成。部分酰胺基团上的C= O 伸缩振动峰向低波数移动,移动到1 648 cm-1处以及2 919 cm-1处时-N-H的伸缩振动峰从双峰分裂成了三峰,这是由于C=O 和-NH2之间形成了氢键造成的。综上所述,证明成功制备了PAESB-L 水凝胶。

图4 PAM,PAS,PAESB-L 水凝胶的全反射红外光谱Fig.4 ATR-IR spectra of PAM, PAS, PAESB-L hydrogel
2.3 水凝胶自修复能力
将用于拉伸力学性能测试的水凝胶样品切成两段后,在37 ℃下,将截面重新连接在一起并静置不同时间(6、24、48 h)重新测量水凝胶的拉伸性能,结果如图5(a)所示。切断的水凝胶自修复6、24、48 h后断裂伸长率分别为252.1%、446.3%、718.3%;自修复48 h 后水凝胶的可拉伸长度大约为初始水凝胶样条的7 倍,自修复效率为56.1%(自修复后和原始水凝胶的断裂伸长率的比值)。由于断裂处的截面被破坏,导致水凝胶的自修复效率没有达到100%[30],但是其自修复后的柔韧性优于相关文献报道[20,31]。由图5(b)可以看到,在室温条件下,断裂后的水凝胶自修复1 h 后,可以承受自身质量而不断裂,红色框内圈出了水凝胶自修复后外部残留的裂痕,可以看到水凝胶自修复后外部裂痕基本消失。

图5 (a)水凝胶试样愈合不同时间的拉伸应力-应变曲线;(b)自修复1 h 后的水凝胶可以承受自身质量而不断裂的照片Fig.5 (a) Tensile stress-strain curves of hydrogel samples healing for different time; (b) Optical demonstration of a healed hydrogel withstanding its own weight after healing 1 h
2.4 水凝胶的力学性能
2.4.1 单体质量比对水凝胶拉伸性能的影响 表1所示为单体质量比不同时水凝胶的力学性能。由表1可以看到,拉伸强度随着mAEAM∶mAM和mASI∶mAEAM的增大而增大,而断裂伸长率随着mAEAM∶mAM和mASI∶mAEAM的增大而减小。水凝胶弹性模量和拉伸强度随着mASI∶mAEAM的增大而增大,而断裂伸长率随着mASI∶mAEAM的增大而减小,当mAEAM∶mAM=1∶12,mASI∶mAEAM= 1∶80 时,水凝胶的拉伸强度(103.6 kPa)和弹性模量(6.0 kPa)达到最小。这主要是因为当mASI∶mAEAM增大时,PAES-L 水凝胶体系中席夫碱的含量增加,导致形成的水凝胶网络更为紧密,抗拉伸能力更强。当mASI∶mAEAM逐渐减少时,水凝胶网络中席夫碱物理交联程度减弱,水凝胶网络组成松散,分子链活动限制较小,聚合物链的可活动空间逐渐变大,断裂伸长率增加[32]。

表1 不同单体质量比的水凝胶的力学性能Table 1 Mechanical properties of hydrogels with different monomer mass ratios
2.4.2 单体总质量分数对水凝胶拉伸性能的影响固定mAM∶mAEAM∶mASI= 960∶80∶1,调节水凝胶中单体总质量分数(wT,即w(ASI)+w(AEAM)+w(AM)),研究单体总质量分数对水凝胶力学性能的影响。图6所示为单体总质量分数不同时水凝胶应力-应变曲线和弹性模量。

图6 单体总质量分数不同时水凝胶的应力-应变曲线(a)和弹性模量(b)Fig.6 Stress-strain curves (a) and elastic modulus (b) of hydrogels with different total mass fractions of monomer
由图6(a)可知,随着单体总质量分数的增加,水凝胶的拉伸强度先变小后变大,断裂伸长率先变大后变小,当单体总质量分数为20.00%时,拉伸强度最小,为101.3 kPa;断裂伸长率最大,可以达到1 589.2%。随着单体总质量分数的增大,聚合物链长增长的同时,交联网络也变得更加密集。聚合物链长的增加使得水凝胶断裂伸长率变大;当水凝胶交联网络变得密集时,水凝胶的抗拉强度增加。
由图6(b)可以看到,当单体总质量分数为20.00%时,水凝胶的的弹性模量最小,为6.0 kPa。这也是因为聚合物链长增长受单体浓度影响较大,在交联网络变得更加密集的同时,链增长使得水凝胶网络愈发柔韧,同等外力作用下产生的形变更大,从而使得水凝胶弹性模量减小;随着水凝胶交联网络变得更加密集,聚合物链长增长受单体浓度的影响逐渐变小,同等外力产生的形变逐渐变小,水凝胶弹性模量增大。
2.4.3 BORAX 用量对水凝胶拉伸性能的影响 为研究BORAX 的用量对PAES-L 水凝胶力学强度的影响,固定单体总质量分数,仅对BORAX 质量分数进行调节。如表2 所示,由于BORAX 是通过水解生成的四羟基硼酸根离子()来影响AEAM 上氨基基团的数量,从而对水凝胶的力学性能造成影响,故以m(BORAX)∶m(AEAM)来表示BORAX 的用量。

表2 BORAX 与AEAM 质量比Table 2 Mass ratio of BORAX and AEAM
图7(a)所示为PAES-L 水凝胶应力-应变随BORAX 和AEAM 的质量比变化而改变的曲线。从图中可以看出:随着m(BORAX)∶m(AEAM)的增加,水凝胶的断裂伸长率先增加后减小,拉伸强度一直增加,当m(BORAX)∶m(AEAM)为36∶200 时,水凝胶的断裂伸长率达到最大值1 280.2%,而拉伸强度也达到300.3 kPa。由于体系中AEAM 的质量被固定,BORAX 与AEAM 的质量比增加只会影响水凝胶体系中基团的去质子化,聚合物链长不受影响,只增加了水凝胶网络中席夫碱和氢键的数量,使得水凝胶交联网络更加密集,限制了水凝胶网络的的弹性形变,所以导致水凝胶断裂伸长率呈现先上升后下降,而拉伸强度持续增大的趋势。由图7(b)可以更清楚地看到随着BORAX 与AEAM 质量比的增加,水凝胶拉伸强度呈上升的趋势。但是水凝胶的弹性模量先下降后上升。这主要是由于在BORAX质量分数较小以及基团的去质子化程度较低时,水凝胶聚合物链段更加灵活,水凝胶在纵向拉伸力的引导下,链与链之间排布愈发规整并发生了取向行为,使得水凝胶具有较高的弹性模量;随着基团的去质子化程度增加,席夫碱和氢键的形成限制了水凝胶中聚合物链的活动,削弱了聚合物链的取向行为,使得水凝胶的弹性模量逐渐降低;当基团的去质子化程度进一步加深,水凝胶网络中充斥着大量的席夫碱与氢键作用时,符合胡克定律的形变需要更大的应力来实现,因此水凝胶弹性模量呈现上升趋势。PAESB-L 水凝胶弹性模量最低可达15.3 kPa,说明此配比下水凝胶受外力产生形变的能力较强,易于形变,这有利于水凝胶在传感领域的使用[31]。

图7 BORAX 与AEAM 质量比不同时水凝胶的应力-应变曲线(a)以及抗拉强度和弹性模量(b)Fig.7 Stress-strain curves (a) and trends of tensile strength and elastic modulus (b) with different m(BORAX)∶m(AEAM)
2.5 水凝胶的导电性能
AEAM 作为一种酸性聚电解质,增强了PAESBL 水凝胶的导电能力。通过电化学工作站,在室温环境下,采用三探针交流阻抗法对各组分水凝胶的电导率进行了测试。图8(a)所示为PAM、PAS、聚(丙烯酰胺-N-丙烯酰氧基琥珀酰亚胺-N-2-氨基乙基丙烯酰胺盐酸盐)(PAES)、聚(丙烯酰胺-N-丙烯酰氧基琥珀酰亚胺-N-2-氨基乙基丙烯酰胺盐酸盐)-硼砂(PAESB)、PAESB-L 水凝胶的交流阻抗曲线,根据散点图在Z’轴的截距可以得到内部电阻Rb,再根据式(3)可得该水凝胶的电导率。由图8(b)可知,PAM、PAS 水凝胶的电导率非常低,分别只有0.081 S/m 和0.089 S/m。但是,加入AEAM 后PAES 水凝胶的电导率可达到1.51 S/m;加入BORAX 后PAESB水凝胶的电导率可达到1.78 S/m;加入LiCl 后PAESB-L 水凝胶的电导率变化较大,可达到3.11 S/m。因此,聚电解质以及电解质的加入使得离子导电水凝胶的电导率得到显著的提升。
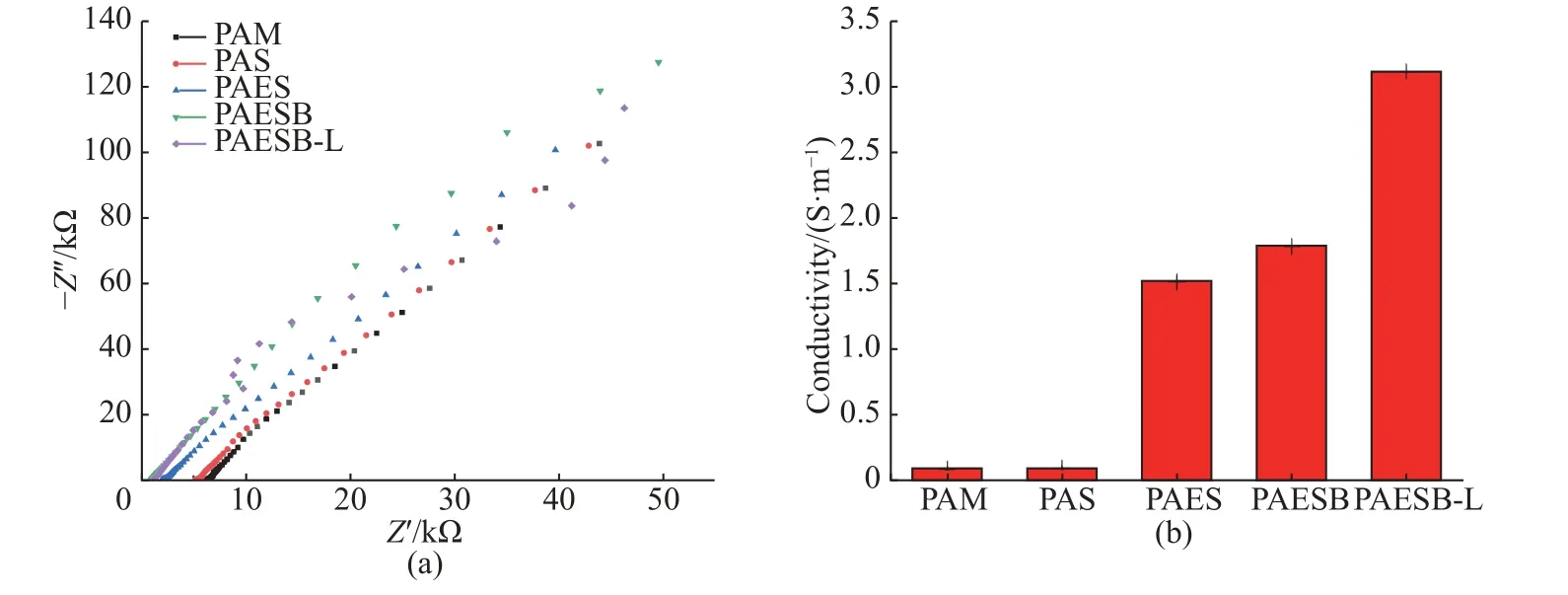
图8 不同组分的水凝胶的交流阻抗曲线(a)和电导率(b)Fig.8 Electrochemical impedance curves (a) and conductivities (b) of hydrogels with different compositions
3 结 论
(1)PAESB-L 水凝胶具有较好的自修复能力,在自修复48 h 后断裂伸长率依旧可以达到718.3%。
(2)PAESB-L 水凝胶具有较高的柔韧性,通过改变各组分质量比,其断裂伸长率最高可达1 495.2%,拉伸强度最高可达328.9 kPa。
(3)PAESB-L 水凝胶具有较高的电导率,在室温环境下,其电导率最高可达3.11 S/m。
猜你喜欢
中学生理科应试(2024年1期)2024-05-18 13:02:52
山东冶金(2023年6期)2024-01-10 01:33:30
理化检验(物理分册)(2017年5期)2017-06-01 11:29:45
钢管(2016年4期)2016-11-10 07:37:00
焊管(2015年4期)2015-12-19 07:01:37
电源技术(2015年12期)2015-08-21 08:58:30
邵阳学院学报(自然科学版)(2015年2期)2015-06-05 12:22:39
原子与分子物理学报(2014年3期)2014-02-28 22:18:23
无机化学学报(2014年7期)2014-02-28 17:32:28
无机化学学报(2014年4期)2014-02-28 17:31:19
