
海分枝杆菌embA 低表达菌株的构建及巨噬细胞侵染实验
2023-11-03 02:40:18李烨雨刘含梅黄滢睿
复旦学报(自然科学版) 2023年5期
李烨雨,刘含梅,黄滢睿,郭 滨,张 鹭
(1.复旦大学 生命科学学院 微生物学与免疫学系,上海 200438;2.复旦大学 遗传工程国家重点实验室,上海 200082;3.复旦大学 基础医学院,上海 200030;4.复旦大学 生命科学学院 实验教学中心,上海 200438)
分枝杆菌具有独特且复杂的细胞壁结构,由共价联接的“核心”结构,分枝菌酸-阿拉伯半乳聚糖-肽聚糖及外层脂类构成[1],具有很低的通透性和流动性,形成有效的渗透屏障,导致分枝杆菌对多数常用抗生素天然耐药[2]。同时在入侵宿主的过程中,细胞壁也发挥了重要的免疫调控和免疫逃避作用[3]。因此,靶向分枝杆菌细胞壁生物合成途径的抗结核药物的开发,以及对于分枝杆菌细胞壁成分的毒力作用机制的研究,一直备受关注。
由emb基因编码的阿拉伯糖基转移酶在分枝杆菌中功能保守,其中EmbA/EmbB糖基转移酶催化α(1→3)Araf的聚集和六阿糖腺苷结构(Afaf6)的形成过程,在细胞壁阿拉伯半乳聚糖(Arabinogalactan,AG)合成过程中发挥重要催化作用[4],也是一线抗结核药物乙胺丁醇的作用靶点[5]。在正常培养条件下,embA/embB都是致病性结核分枝杆菌(Mycobacteriumtuberculosis,M.tb)的必需基因,embA上游存在功能性启动子区域,可独立于上游embC基因,与下游embB共转录。embA基因启动子活性在整个生长阶段和应激处理后保持不变,但在缺氧诱导的持留期显著降低[5]。在快生长的耻垢分枝杆菌(Mycobacteriumsmegmatis,M.smegmatis)中,embA缺失突变会造成细胞壁AG 合成缺陷,导致细菌形态改变、耐酸性轻微丧失、疏水性和抗生素敏感性增加等[5]。表明了embA基因对分枝杆菌生理功能的广泛影响。海分枝杆菌(Mycobacteriummarinum,M.marinum)是鱼类和变温动物结核病样疾病的病原体,也是人类的条件性致病菌,与M.tb的embA基因序列相似性为85.65%,二者AG 生物合成过程一致。但在M.tb和M.marinum等致病性分枝杆菌中,embA对细菌毒力的影响,尚缺乏实验证据。
本研究首先评估了海分枝杆菌参与AG合成基因的本底表达水平,并通过CRISPRi基因编辑技术构建embA低表达菌株,在巨噬细胞感染模型中评估突变株的细胞毒力。在宿主识别病原菌的过程中,髓样分化因子88(Myeloid Differentiation factor 88,MyD88)参与的免疫反应尤为关键,影响多种免疫因子分泌[6-10]。因此,进一步探究embA低表达株对巨噬细胞MyD88表达的影响,以及MyD88下游促炎症细胞因子和趋化因子的表达情况,为解析致病性分枝杆菌中embA基因对细菌毒力的影响提供参考和实验支持。
1 材料和方法
1.1 材料
海分枝杆菌MycobacteriummarinumM 菌株、glfT2低表达海分枝杆菌、小鼠巨噬细胞系J774A.1和人类单核细胞系THP-1由本实验室保存;CRISPRi骨架质粒PLJR962由加拿大多伦多大学刘军教授实验室惠赠;大肠杆菌EscherichiacoliDH5α感受态菌株购自北京全式金生物技术有限公司。
1.2 试剂
DifcoTMMiddlebrook 7H9 Broth 购自BD Biosciences 公司;ADC 增菌液(Album in of bovine-Dextrose-Catalase,包括牛血清白蛋白50g,过氧化氢酶0.03g,葡萄糖20g,定容至1L后0.22μm 滤膜抽滤除菌);OADC增菌液(Oleic acid-Album in of bovine-Dextrose-Catalase,包括牛血清白蛋白50g,过氧化氢酶0.05g,葡萄糖: 20g,油酸: 0.5g,NaCl: 8.5g,定容至1L后0.22μm 滤膜抽滤除菌);BsmBI限制性核酸内切酶及酶切缓冲液购自NEW ENGLAND BioLabs公司;PrimeScript RT reagent Kit with gRNA Eraser 购自TaKaRa 公司;Talent qPCR PreMix(SYBR Green)购自Tiangen 公司;TRIzol Reagent购自Ambion公司;分枝杆菌抗酸染色液购自珠海贝索生物技术有限公司;Dulbecco's Modified Eagle Medium(DMEM)培养基,Roswell Park Memorial Institute(RPMI) 1640培养基,胎牛血清(FBS),Penicillin-Streptomycin双抗和0.25%胰酶购自Gibco公司;台盼蓝染色液购自碧云天公司;MyD88 Rabbit mAb,GAPDH Rabbit mAb 和Anti-rabbit lgG HRP-Linked购自Cell signaling 公司;SuperSignalTMWest Femto Maxium Sensitivity Substrate购自Thermo公司;佛波酯(Phorbol 12-Myristate 13-Acetate,PMA)购自MedChemExpress公司;Phalloidin-iFluor 647试剂和DAPI染色试剂购自Abcam 公司。
1.3 方法
1.3.1 海分枝杆菌培养
在7H9无抗培养基(7H9粉末4.7g,甘油2mL,定容至900mL,121℃高温灭菌10min后,冷却至50~60℃,加入100mL ADC增菌液)中复苏海分枝杆菌,30℃避光震荡培养至OD600为1.0左右,传代活化到平台期。按照2‰接种量接种到无菌7H9液体培养基中,30℃震荡培养至对数期用于后续实验。
1.3.2 qRT-PCR 检测基因相对表达量
离心收集不同生长阶段的海分枝杆菌菌体,TRIzol法提取RNA,使用TaKaRa 的逆转录试剂盒逆转录RNA。qPCR所用的引物如表1所示,内参基因选用sigA,用相对定量的方法测定目标基因的表达水平。

表1 实时荧光定量PCR 引物Tab.1 Primers for real-time fluorescence quantification
1.3.3 海分枝杆菌embA低表达菌株构建及基因表达验证
利用CRISPRi法构建低表达菌株。由擎科生物公司合成编码靶向embA基因的sgRNA 的DNA 片段上下游引物序列(上游引物: GGGAGCCGCGGGCACCCTGCCCCCG;下游引物: AAACCGGGGGCAGGGTGCCCGCGGC),退火处理形成DNA 双链片段。使用BsmBI限制性核酸内切酶,55℃单酶切PLJR962质粒。回收酶切产物后用T4DNA 连接酶室温连接退火片段。酶连后转化50mLE.coli感受态细胞,在含50μg/mL 卡那霉素的LB 固体培养基上筛选阳性克隆送测序,测序引物序列为5'-TTCCTGTGAAGAGCCATTGATAATG-3'。
测序正确的质粒以2.5kV,1kΩ 电阻和25μF电流容量电转入海分枝杆菌感受态细胞中。构建成功的PLJR962质粒上含有CRISPi元件,在分枝杆菌中以质粒形式稳定存在,无水四环素(Anhydro-tetracyline,ATc)能够通过Tet-on表达调控系统抑制目标基因的转录[11]。将生长到OD600=1的低表达菌株接种到50mL 7H9培养基(含25μg/mL卡那霉素)中,稀释至初始OD600=0.05,加入ATc 诱导(200ng/mL),30℃摇床震荡培养。在OD600=1.0或2.0时收集菌体提取RNA,参照1.3.2节的方法测定embA等基因相对表达量。
1.3.4 抗酸染色
取对数生长中期(OD600=2.0)菌液适当稀释,均匀涂布在载玻片上,菌液干燥固定后,依次滴加石碳酸复红染液染色,酸性酒精溶液脱色,亚甲蓝溶液复染。干燥后置于光学显微镜(Leica)下观察细菌形态,并用Image J比较菌体的长度l。
1.3.5 巨噬细胞侵染实验
在DMEM 培养基(含10% FBS,100units/mL Penicillin,100μg/mL Streptomycin)中37℃传代扩增培养小鼠J774A.1巨噬细胞系。或在1640培养基中(含10%FBS,100units/mL Penicillin,100μg/mL Streptomycin)复苏THP-1细胞系,传代培养后用200nmol/L PMA诱导24h贴壁分化成巨噬细胞,继续培养48h。取对数生长中期的embA低表达株及对照株菌液,用胰岛素针反复吹打制备单细菌悬液。按照感染复数(Multiplicity Of Infection,MOI)=5的比例,用加入ATc(200ng/mL)的DMEM(不含FBS)稀释菌悬液,加入巨噬细胞培养孔侵染。
1.3.6 台盼蓝染色测定J774A.1细胞存活率
台盼蓝染色是测定细胞存活率的常用方法之一,按1.3.5节的方法侵染J774A.1巨噬细胞24h后,吸弃每孔细胞上清,用PBS润洗细胞一遍,每孔(12孔细胞培养板)加入100μL 0.25%胰酶消化1min,再加入等体积的含FBS的DMEM 培养基。吹打混匀后吸取细胞液,加入等体积台盼蓝染色液计数活细胞数和总细胞数,计算细胞存活率Rsurvival。
1.3.7embA低表达株在J774A.1胞内CFU 计数
按1.3.5节方法,以加入侵染液的时刻作为侵染0h,侵染J774A.1巨噬细胞24h后弃掉细胞培养上清,加入0.1% Triton X-100,室温消化细胞10min,吹打混匀后用无菌PBS缓冲液系列稀释后涂布到7H10固体培养基上(7H10粉末19g,甘油5mL,定容至900mL,121℃高温灭菌10min后冷却至50~60℃,加入100mL OADC增菌液。添加25μg/mL 卡那霉素),每组菌每个稀释梯度涂布3个平行板,30℃培养箱倒置培养约10d计算板上菌落形成单位(Colony Forming Units,CFU)数量Ncolony。
1.3.8 巨噬细胞骨架蛋白F-actin细胞免疫荧光染色观察
海分枝杆菌侵染巨噬细胞3h后,吸弃细胞培养液。一部分加入100μL稀释的Phalloidin-iFluor 647试剂,室温避光孵育1h,PBS润洗后DAPI染色液染色15min,换上PBS置于荧光显微镜下(Olymbus BX63)观察。另一部分换上含有FBS和双抗的1640培养基培养(含ATc 200ng/mL),同样操作处理。每组3个平行重复。
1.3.9 MyD88蛋白表达水平验证及细胞因子分泌检测
海分枝杆菌侵染J774A.1细胞12h后,移走细胞培养上清,余下每孔(6孔板)加入200μL细胞裂解液,室温消化后测定蛋白浓度。蛋白样在SDS-PAGE 胶电泳后采用“夹心法”转PVDF 膜,稀释MyD88 Rabbit mAb,4 ℃过夜孵育;用抗兔lgG-HRP 二抗(1∶10000)孵育1h,1×TBST 溶液洗膜后使用SuperSignalTMWest Femto Maxium Sensitivity Substrate显色液显色。比较野生株和embA低表达株侵染后细胞MyD88蛋白表达水平。以GAPDH 为内参蛋白,同步操作并显色。
海分枝杆菌侵染J774A.1细胞18h后,收集巨噬细胞培养上清并测定蛋白浓度。使用液相悬浮式蛋白芯片检测上清中免疫因子浓度,共检测了23种常见细胞因子和趋化因子。样本检测在上海柏辰生物科技有限公司完成。
2 结果
2.1 海分枝杆菌不同生长阶段中AG 合成相关基因本底表达水平
在海分枝杆菌野生株(Wild Type)对数生长前期(OD600=1.0)和中期(OD600=2.0)时提取细菌RNA,qRT-PCR检测7个参与AG 合成的关键基因(aftA/aftC/aftD/embA/embB/glfT1/glfT2)表达水平。如图1所示,与对数生长前期相比,基因embA和glfT2在对数中期表达显著上调(P<0.05),上调倍数分别为1.4和1.6倍,其他基因的相对表达量则无显著差异。

图1 野生型海分枝杆菌AG 合成基因相对表达量Fig.1 Relative expression of AG biosynthesis genes in wild type M.marium
2.2 embA 低表达的海分枝杆菌突变株的验证及形态比较
embA为分枝杆菌生长必需基因[12],完全敲除会导致菌体无法生长,因此构建低表达株是更为可行的策略。构建CRISPRi敲低载体,电转化海分枝杆菌后ATc 诱导表达dCas9 蛋白,在sgRNA 的靶向作用下dCas9蛋白阻止了embA基因的转录,从而低表达embA,基因表达水平被抑制的程度取决于CRIPRi元件上的sgRNA 的特异性。我们检测了含相同质粒的3 个不同单克隆,证明embA基因表达水平一致,敲低骨架质粒系统工作效率稳定,取同一株单克隆开展后续实验。qRT-PCR 实验比较突变株与野生株中embA基因相对表达量,图2(a)显示对数生长前期和中期低表达株中embA表达分别下调50%(P<0.001)和75%(P<0.001)。结合图1野生型细菌中embA基因本底表达结果,证明本研究的embA低表达突变株株构建成功(菌株命名为EmbA_KD)。本实验室前期用同一方法成功构建了glfT2低表达菌株,GlfT2与EmbA 均参与分枝杆菌AG 合成,但GlfT2催化AG 半乳糖链的合成。本研究将这一glfT2低表达株作为参照之一(菌株命名为GlfT2_KD),以评估embA低表达与突变株细菌表型及细胞毒力变化之间的相关性。后续感染实验中,同步检测这两株突变株中对应基因转录水平,确保基因持续稳定表达。

图2 embA 低表达(EmbA_KD)海分枝杆菌的验证及形态比较(400×)Fig.2 Validation of embA low-expression mutant and morphological comparison (400×)
利用抗酸染色观察EmbA_KD 株细菌形态变化。如图2(b)所示,细菌为红色,呈典型的杆状特征,各组菌体形态无明显差别。通过统一标尺及图像处理软件Image J比较菌体长度,野生株及EmbA_KD、GlfT2_KD 突变株的菌体长度分别为(180.6±5.979)、(161.5±11.47)和(163.0±12.43)μm,无统计学意义上的差异。
2.3 EmbA_KD株中AG 合成基因相对表达水平
在对数生长前期和中期,分别比较EmbA_KD 和野生株中参与AG 合成关键基因(aftA/aftC/aftD/embA/embB/glfT1/glfT2)的相对表达水平。如图3 所示,在对数生长前期,EmbA_KD 中embA基因低表达50%(图3(a)),aftD基因表达显著上调了2.2倍(P<0.0001)。而在对数生长中期,EmbA_KD 相比于野生株所有基因均显著下调45%~80%(P<0.0001)(图3(b))。
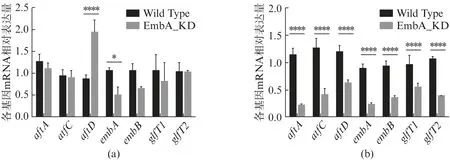
图3 EmbA_KD 海分枝杆菌中AG 合成基因的相对表达量Fig.3 Relative expression of AG biosynthesis genes in EmbA_KD
2.4 巨噬细胞感染模型评估EmbA_KD的毒力变化
利用海分枝杆菌细胞感染模型来评估embA基因下调对细菌毒力的影响。以MOI=5 侵染小鼠J774A.1巨噬细胞24h后,台盼蓝染色显示EmbA_KD 株侵染组细胞存活率为(48.69±1.47)%,显著高于对照株(27.02±1.70)%和GlfT2_KD 株(32.14±2.89)%侵染组(P<0.05),细胞存活率的提高初步表明embA的低表达导致了海分枝杆菌毒力降低。同步检测了被感染细胞内细菌CFU 变化,如图4(b)所示,侵染24h后EmbA_KD 株的胞内活菌数为(2.15±0.15)×106CFU,显著高于对照株(0.89±0.08)×106CFU 和GlfT2_KD 株(1.04±0.10)×106CFU。
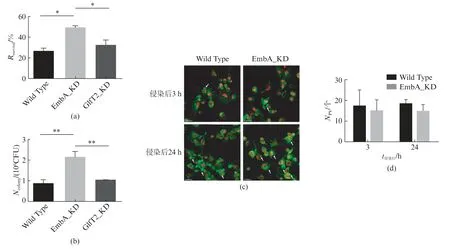
图4 巨噬细胞感染模型测定EmbA_KD 株毒力变化Fig.4 Virulence changes of EmbA_KD determined by macrophage infection model
人源巨噬细胞对分枝杆菌的吞噬杀伤作用强于小鼠巨噬细胞,且被感染时不易发生细胞死亡。本研究选用人源巨噬细胞进一步评估EmbA_KD株的毒力变化。当巨噬细胞被侵染时,通过细胞骨架重排形成吞噬小泡(Phagocytic Vesicle,PV)吞噬海分枝杆菌,后续与溶酶体融合杀伤细菌。免疫荧光染色观察微丝肌动蛋白F-actin细胞骨架重排情况,发现侵染3h和24h后,野生株和EmbA_KD株侵染组细胞内均有点状或小泡状聚集的吞噬小泡形成(见 第590页,图4(c)白色箭头指向)。计数全部视野下所有细胞吞噬小泡,计算平均吞噬小泡数量与细胞数量比值,如图4(d)所示,3h和24h时间点下两组被侵染细胞间无显著差异。
2.5 J774A.1巨噬细胞侵染后MyD88表达量及细胞因子分泌量检测
通过巨噬细胞侵染模型评估embA低表达菌株相关的细胞免疫反应。将野生株、EmbA_KD 和GlfT2_KD 株分别以MOI=5侵染小鼠J774A.1巨噬细胞12h后,Western blot检测细胞内MyD88的蛋白水平,如图5(a,b)所示,EmbA_KD 株侵染后巨噬细胞中MyD88蛋白表达量相比野生株和GlfT2_KD 株侵染组分别显著上调1.97倍(P<0.0001)和1.60倍(P<0.001)。
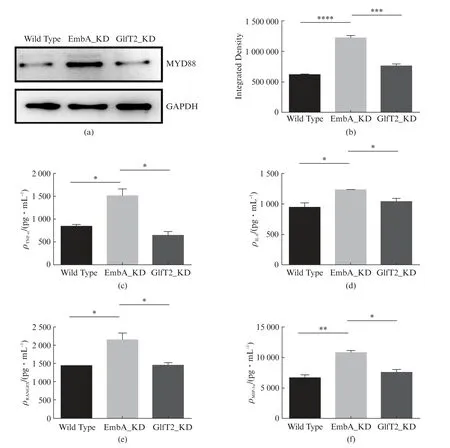
图5 J774A.1巨噬细胞系被侵染后MyD88表达量及细胞因子表达水平Fig.5 MyD88 expression level and cytokines level of J774A.1 macrophage line 12h post infection
因为细胞因子分泌具有一定的延时性,在侵染18h后使用液相悬浮式蛋白芯片检测细胞上清中常见细胞因子与趋化因子浓度,重点关注MyD88参与调控的下游细胞因子浓度变化,如TNF-α和IL-6。
图5(c,d)表明,EmbA_KD 株侵染后巨噬细胞分泌更高水平的TNF-α,较野生株和GlfT2_KD 株侵染的细胞分别上调1.77倍(P<0.05)和2.15倍(P<0.05);同样IL-6水平较两者各上调1.31倍(P<0.05)和1.18倍(P<0.05)。
受到MyD88调控的趋化因子RANTES(Regulated upon Activation Normal T cell Expressed and Secreted factor,也称CCL-5)、巨细胞炎性蛋白MIP-1α(也称CCL-3)也同样显著上调,如图5(e,f)所示,embA低表达株侵染巨噬细胞后,较野生株侵染组上调1.49 倍(P<0.05)和1.58 倍(P<0.01);较GlfT2_KD 株侵染组上调1.47倍(P<0.05)和1.43倍(P<0.05)。图5同时显示,野生株和GlfT2_KD株侵染巨噬细胞后,胞内MyD88蛋白表达和细胞因子分泌量均无显著差异。表明MyD88表达和细胞因子分泌的变化是由embA低表达菌株导致。
3 讨论
AG 是分枝杆菌细胞壁的关键成分,在细菌不同生长阶段可能需要量不同,取决于新合成的细胞壁的数量。AG 生物合成的变化与其合成通路中相关蛋白的表达或活性直接相关。因此,本研究首先测定了海分枝杆菌野生株在对数生长前期、中期,7个AG 合成基因的转录水平变化。embA和glfT2在对数中期转录水平升高,提示这两个基因更易于受到细菌生长状态的影响。利用CRISPRi技术构建了embA低表达株,证明在对数生长前期和中期,embA的表达分别下调了50%和75%,延长培养时间,表达水平维持稳定。同步诱导处理对照菌株和GlfT2_KD株,embA表达水平不变,可以排除ATc 诱导敲低的时间累积效应。有研究表明,在M.tb和M.smeg中,embA和embB存在共转录关系[12]。虽然在M.marinum中未见直接研究报道,但M.marinum基因组中embB紧邻embA片段下游,embA低表达株中embB基因同步显著下调,表明这两个基因同样存在共转录关系。除了embB,EmbA_KD突变株中,aftA/aftC/aftcD/gflT1/glfT2这些参与AG 合成的基因的表达虽然也出现下降,但难以确定是embA的变化导致,因为在基因组中,除了aftA位于embA基因上游近端,aftC/aftcD/gflT1/glfT2基因座位距离embA都非常远,推测可能是存在某种反馈调节机制,如细胞壁成分变化导致这些基因表达受到反馈抑制,但有待实验验证。
分枝杆菌进入宿主后首先被巨噬细胞吞噬,细菌会在早期的吞噬体内存活甚至复制,并且抑制吞噬溶酶体的成熟,以逃避免疫清除;一旦穿透吞噬体膜并泄漏到胞浆中,分枝杆菌可以复制并导致感染细胞坏死,释放出大量的细菌,进而感染临近的细胞[13]。虽然关于分枝杆菌在巨噬细胞质中复制的细节尚不清楚,但在吞噬过程中细胞骨架的组装及解聚发挥主要作用,肌动蛋白聚合及内吞噬小泡形成是吞噬作用的必要条件。本研究发现,EmbA_KD 被THP-1巨噬细胞吞噬3h及24h后,内吞噬小泡形成及细胞骨架重排情况与野生株相比并无明显差别,可见海分枝杆菌中embA表达量下降不会影响海分枝杆菌被吞噬细胞吞噬的生理过程。进一步探索EmbA_KD 株的毒力变化,发现侵染24h后,巨噬细胞内EmbA_KD活菌数量高于其他两组,而被感染细胞存活率也最大。当比较胞内细菌量和存活细胞的比值时,各组之间并没有显著差异,表明EmbA_KD株侵染的细胞更容易存活,embA低表达导致细菌的细胞毒力下降。后续可以在海分枝杆菌的天然宿主斑马鱼感染模型中进一步验证embA低表达对分枝杆菌毒力的影响。
靶向TLR 信号是胞内菌和病毒普遍采用的一种免疫逃避策略。分枝杆菌通过胞内表达ESX 分泌系统成分和各种糖脂等毒力因子,改变正常宿主细胞信号通路,其中大多数是针对MyD88依赖的信号级联反应。MyD88作为Toll样受体的关键配体蛋白,能够激活MAPK 及NF-κB信号通路从而引起宿主对结核杆菌的天然免疫反应,影响促炎症细胞因子如TNF-α、IL-6,趋化因子RANTES、MIP-1α 等的分泌[6-10]。MyD88的缺失会导致细胞对分枝杆菌易感性的增加和宿主体内炎症反应的减弱[7,14]。被BCG感染的骨髓间充质干细胞MyD88表达显著上调[15],临床研究也显示活动性结核病人的血液转录组中MyD88表达量上升[16]。细菌多糖或脂多糖、外毒素、细胞因子或者microRNA 都能够调控MyD88表达量[17-19],为了探索EmbA_KD 株细胞毒力的下降是否与MyD88 及其下游免疫因子相关,本研究在J774A.1小鼠巨噬细胞感染模型中发现,当AG 合成相关基因普遍下调的embA低表达株侵染细胞时,MyD88蛋白表达显著上升,引起下游促炎症细胞因子TNF-α和IL-6水平的增加。细胞因子是分枝杆菌免疫反应的重要效应子和调控子,致病性和非致病性分枝杆菌的研究表明,分枝杆菌的毒力与促炎性细胞因子的分泌呈负相关[9-10]。本研究结果从关键免疫分子角度证明了embA低表达导致细菌的毒力下降。MyD88依赖的NF-κB等信号通路对巨噬细胞RANTES和MIP-1α的分泌也至关重要[9-10],本研究检测到这两种趋化因子的上调,提示EmbA_KD 株侵染后,巨噬细胞对单核细胞和粒细胞等趋化募集作用增强[20-21]。值得关注的是,作为参照的glfT2低表达株侵染巨噬细胞并没有引起这一现象,说明MyD88的高表达和细胞因子分泌水平的上升主要与embA基因的表达下调相关。
本研究发现EmbA_KD 株在对数中期时AG 合成通路中其他6个基因也普遍下调,提示AG 表达水平或AG 成分可能发生变化。分枝杆菌细胞壁中的AG 具有很强的免疫刺激和调控潜力,常用作抗结核疫苗的免疫佐剂[22]。AG 可能激活MAPK/ERK/NF-κB 等信号通路,诱导巨噬细胞炎症反应引起肺部损伤[23]。EmbA_KD 株造成的巨噬细胞MyD88表达量的变化也可能与细胞壁中AG 或其他糖脂表达量发生改变有关,这是一个值得深入探索的现象。
总之,embA低表达导致海分枝杆菌毒力下降,同时引起宿主促炎症细胞因子和趋化因子分泌增强,如果据此改造致病性分枝杆菌,降低或消除细菌毒力的同时提高其免疫原性,有望获得兼具安全性和保护性效果的候选疫苗。而在M.tb或其他致病性分枝杆菌中解析embA等AG 合成相关基因的毒力调控作用,有助于对结核菌细胞壁多糖的深入了解,也有望为抗结核治疗提供新的候选药物靶标。
猜你喜欢
当代水产(2022年1期)2022-04-26 14:35:30
数学物理学报(2022年2期)2022-04-26 14:08:06
云南化工(2021年6期)2021-12-21 07:31:04
新世纪智能(数学备考)(2021年9期)2021-11-24 01:14:34
新世纪智能(数学备考)(2020年9期)2021-01-04 00:25:12
农药科学与管理(2019年6期)2019-11-23 08:17:12
中学生数理化·高一版(2018年10期)2018-11-08 11:06:56
西南农业学报(2016年5期)2016-05-17 05:42:33
西南农业学报(2016年6期)2016-04-16 05:12:51
华南农业大学学报(2015年5期)2015-12-04 03:04:38
