
干灰-离子色谱法测定食品中氯化物含量的研究
2023-10-27 09:21蔡志兴
福建轻纺 2023年10期
蔡志兴
(福建省福食安检测技术有限公司,福建 泉州 362200)
目前我国食品中氯化物的测定采用GB 5009.44—2016《食品安全国家标准 食品中氯化物的测定》中的间接沉淀滴定法、电位滴定法、直接沉淀滴定法,其中间接沉淀滴定法和直接沉淀滴定法容易受样品颜色干扰,不适用于深颜色食品中氯化物的测定[1-2]。离子色谱法具有操作简单,灵敏度高和准确度高、且对环境友好等特点[3-4],目前广泛应用于各类产品中各种阴离子的测定[5-7]。
刘美芹、孙雪萍等人先后采用直接超声提取离子色谱上机检测方式对畜禽肉、水产品等食品进行氯化物含量的测定[4,8]。干灰法前处理可有效去除食品中其他组分,能有效提取出产品中氯化物,有效避免其它杂质的干扰及对色谱柱损害,本文构建干灰-离子色谱法对食品中氯化物含量进行测定。
1 实验试剂与仪器
1.1 试剂与设备
试剂:氯离子标准溶液1000 mg/L(坛墨质检科技股份有限公司);超纯水;50%氢氧化钠溶液[赛默飞世尔科技(中国)有限公司].
仪器:离子色谱仪ICS-600、DS5电导检测器、阴离子抑制器(Dionex AERS 500 Carbonate),[赛默飞世尔(上海)仪器有限公司];AS19阴离子分析柱,4×250 mm[赛默飞世尔科技(中国)有限公司];AG19阴离子保护柱,4×50 mm[赛默飞世尔科技(中国)有限公司];50 μL定量环;LS220A电子天平(上海甜美天平仪器有限公司);箱式电阻炉(上海齐欣科学仪器有限公司);电炉。
1.2 色谱条件
淋洗液氢氧化钠浓度20 mmol/L,流速1.0 mL/min;电流50 mA,进样量50 μL,采样时间16 min。
1.3 标准曲线的制作
准确吸取氯离子标准溶液(1000 mg/L)并且依次稀释并配制为0.0、0.50、1.0、2.0、5.0、10.0 mg/L的标准工作溶液。
1.4 样品前处理
称取经粉碎制样均匀的肉类制品、蔬菜、水产品、调味料等各类食品0.5 g于坩埚中,用电炉加热碳化后,放入550 ℃马弗炉灰化1 h。冷却后,灰分用80 ℃热水溶解后,转移定容至100 ml,混匀;用滤纸过滤,取滤液1.00 ml定容至100 mL,用0.45μm水性滤膜针头滤器过滤,收集滤液到进样瓶,待测。
2 结果与讨论
2.1 淋洗液的选择
分别以10~20 mmol/L的氢氧化钠为淋洗液进样,结果表明,氯化物都能有效的与杂质分离,综合考虑杂质的干扰峰、分析时间等因素,选择20 mmol/L的氢氧化钠为淋洗液。该条件下样品分离度高,峰形较好,氯化物保留时间约为6.7 min。氯离子标液和样品检测色谱图见图1、2。
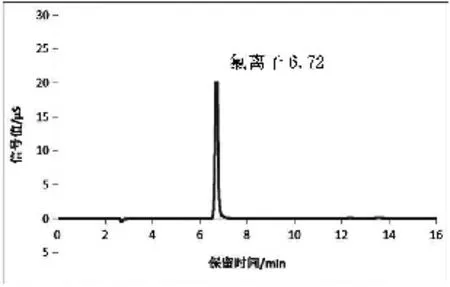
图1 氯离子标液色谱图
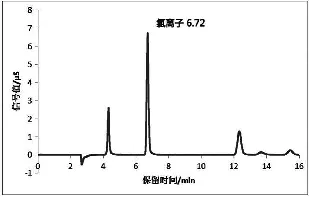
图2 样品检测色谱图
2.2 线性范围和检出限
精确吸取氯离子标准溶液(1000 mg/L)依次稀释并配制为0.0、0.50、1.0、2.0、5.0、10.0 mg/L的系列标准溶液,按上述色谱条件测定峰面积。根据测定结果,得出氯离子回归的回归标准曲线为:y=0.752x-0.055,相关系数r=0.999435,线性关系良好。
根据国际纯粹与应用化学联合会(IUPAC)相关规定,以仪器的3倍信噪比(S/N=3,LOD)计算检出限,本方法中氯离子的仪器最低检出限(LOD)为0.00091 mg/L。按样品称样质量0.5 g计算,以仪器的10倍信噪比计算定量限,本方法食品中氯化物的定量限为0.06 g/kg。
2.3 精密度
选择低、中、高氯离子浓度(0.5、5.0、10.0 mg/L) 3种样品溶液,分别平行进样6次测定,检测结果的相对标准偏差分别为3.48%、1.82%、2.67%。相对标准偏差都在允许的范围内,表明本方法的测定结果有良好的重现性。
2.4 回收率实验
向样品中分别加入 0.5、2.0、9.0 mg/L低中高三种氯离子加标浓度,测试结果显示,加标回收率在96.9%~103.3%,准确度较高(加标回收率结果见表1)。
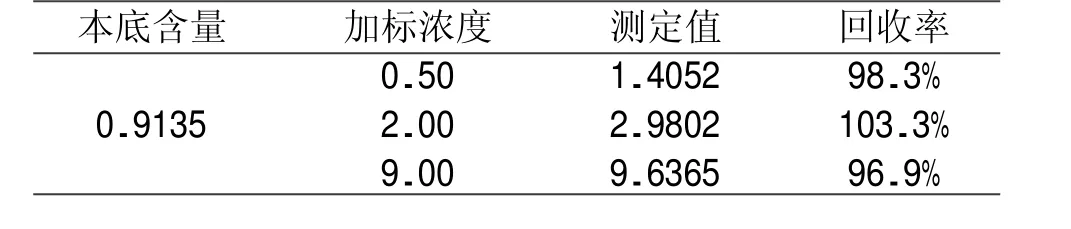
表1 加标回收率结果单位:mg/L
2.5 比对实验
采用本方法与国标GB 5009.44—2016方法进行比对实验,对5份不同的食品样品进行测定,检测结果见表2,对所得结果进行配对t检验,二者差异无统计学意义(t=0.3704,P>0.05)
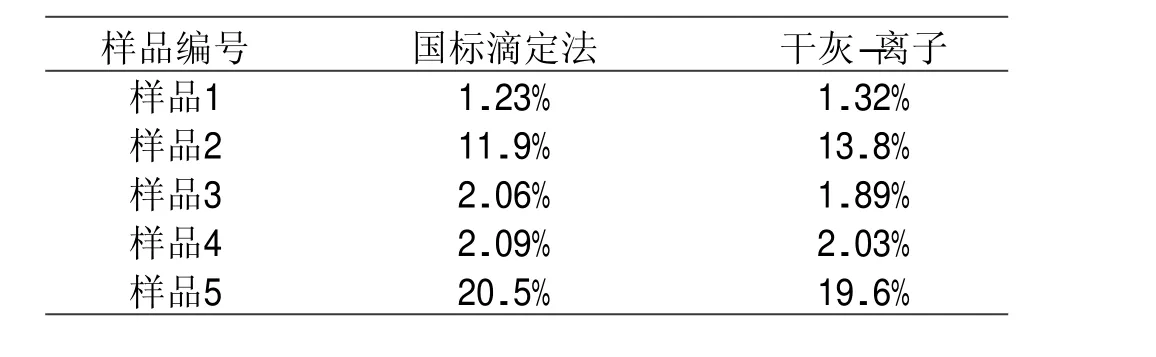
表2 两种方法对照检测结果(以Cl计)
3 小结
本文构建了干灰-离子色谱法测定食品中氯化物含量的方法,考察了方法的精密度和回收率,同时通过与国标方法进行比对实验,结果表明该方法能有效提取出产品中氯化物,与国标中规定方法测定的结果没有显著性差异,并且能有效避免其它杂质的干扰及对色谱柱损害,方法灵敏便捷,响应好,线性范围宽,适用于大批量食品中的氯化物检测。
猜你喜欢
云南化工(2021年11期)2022-01-12
食品安全导刊(2021年20期)2021-11-28
中学生数理化(高中版.高考理化)(2020年3期)2020-05-30
表面工程与再制造(2019年1期)2019-05-11
厦门理工学院学报(2016年1期)2016-12-01
浙江大学学报(工学版)(2016年2期)2016-06-05
河北地质(2016年2期)2016-03-20
物理化学学报(2015年5期)2015-02-28
建筑材料学报(2015年3期)2015-02-28
湿法冶金(2014年3期)2014-04-08
