
BnaSD.C3 is a novel major quantitative trait locus affecting semidwarf architecture in Brassica napus L.
2023-10-16 01:32:40WANGXiaodongCAlYingPANGChengkeZHAOXiaozhenSHIRuiLlUHongfangCHENFengZHANGWeiFUSanxiongHUMaolongHUAWeiZHENGMingZHANGJiefu
Journal of Integrative Agriculture 2023年10期
WANG Xiao-dong ,CAl Ying ,PANG Cheng-ke ,ZHAO Xiao-zhenSHI RuiLlU Hong-fang,CHEN FengZHANG WeiFU San-xiongHU Mao-longHUA Wei,ZHENG Ming#,ZHANG Jie-fu#
1 Provincial Key Laboratory of Agrobiology/Institute of Industrial Crops,Jiangsu Academy of Agricultural Sciences/Key Laboratory of Cotton and Rapeseed,Ministry of Agriculture and Rural Affairs,Nanjing 210014,P.R.China
2 Oil Crops Research Institute,Chinese Academy of Agricultural Sciences/Key Laboratory of Biology and Genetic Improvement of Oil Crops,Ministry of Agriculture and Rural Affairs,Wuhan 430062,P.R.China
Abstract Plant height is a key plant architectural trait that affects the seed yield,harvest index and lodging resistance in Brassica napus L.,although the genetic mechanisms affecting plant height remain unclear. Here,a semi-dwarf mutant,df34,was obtained by ethyl methanesulphonate-induced mutagenesis. Genetic analysis showed that the semi-dwarf phenotype is controlled by one semi-dominant gene,which was located on chromosome C03 using a bulked segregant analysis coupled with whole-genome sequencing,and this gene was named BnaSD.C3. Then BnaSD.C3 was fine-mapped to a 297.35-kb segment of the “Darmor-bzh” genome,but there was no potential candidate gene for the semi-dwarf trait underlying this interval. Furthermore,the interval was aligned to the Zhongshuang 11 reference genome. Finally,combining structural variation analysis,transcriptome sequencing,phytohormone analyses and gene annotation information,BnaC03G0466900ZS and BnaC03G0478900ZS were determined to be the most likely candidate genes affecting the plant height of df34. This study provides a novel major locus for breeding and new insights into the genetic architecture of plant height in B.napus.
Keywords: Brassica napus L.,fine mapping,phytohormone analysis,plant height,transcriptome analysis
1.Introduction
Rapeseed (BrassicanapusL.,AACC,2n=38) is an important oil crop worldwide,which provides approximately 13% of global oil production (Liuet al.2022). Owing to increasing labor shortages and climate issues,the planting areas of the main rapeseed-producing regions,such as China,Europe and Austria,have been decreasing (Zhenget al.2022). It is important to breed cultivars that are suitable for machine production and resistant to abiotic stresses. The utilization of hybrid rapeseed has increased the rapeseed yield in China,but the plant height (PH) of hybrid rapeseed has also increased significantly (Fu and Zhou 2013),which negatively influences lodging resistance. Therefore,new dwarfB.napusgermplasms are urgently needed.
The utilization of dwarf varieties and suitable cultivation methods greatly increased the yields of rice,wheat,and maize during the 1960s and 1970s,resulting in the “Green Revolution” (Penget al.1999). PH is mainly regulated by the biosynthesis and signal transduction pathways of phytohormones. The “Green Revolution”-associated genes of rice and wheat,sd1andRht-1,respectively,are involved in the biosynthesis and signal transduction of gibberellins (GAs) (Penget al.1999;Sasakiet al.2002).Strigolactone has also been found to affect PH and branch number (Jianget al.2013;Zhenget al.2020). Moreover,the biosynthesis and signaling transduction of auxin and brassinosteroids (BR) also affect plant architecture and yield (Korasicket al.2014;Yanget al.2021).
Some dwarf mutants of rapeseed have been obtained,and the genes have been introduced into rapeseed lines to increase the lodging resistance and yield performance(Foissetet al.1995). Quantitative trait loci (QTLs)associated with PH (Luoet al.2015;Wanget al.2015,Wang X Det al.2016),branch angle (Wang Het al.2016;Shenet al.2018) and branch number have also been obtained in rapeseed (Luoet al.2015;Heet al.2017). Liuet al.(2010) identified the semi-dwarf rapeseed mutantds-1,and a substitution in the VHYNP motif of a DELLA protein affects the transduction of the GA signal. With the completion ofB.napusgenome sequencing (Chalhoubet al.2014;Songet al.2020),a growing number of genes that control the PH of rapeseed have been cloned.Liet al.(2019) isolated the dwarf mutantscaby ethyl methanesulfonate (EMS)-induced mutagenesis,and a substitution (glycine-to-glutamic) in the conserved degron motif GWPPV of BnaA3.IAA7 repressed the degradation of the protein,resulting in reduced internode lengths inscaplants.
The development of gene-editing technology has provided a powerful tool for creating new germplasms.For example,Fanet al.(2021) obtained a rapeseed mutant with a semi-dwarf phenotype and compact plant architecture by gene editing,and Zhenget al.(2020) obtained the first rapeseed germplasm with increased branching and reduced PH. Although some dwarf germplasms have been isolated and the genes responsible for plant architecture have been mapped(Wanget al.2020;Yanget al.2021;Yeet al.2022),most of these genes had negative influences on other traits,such as increases in branch angles,and reductions in silique number and disease resistance. Consequently,only a few genes have been applied in rapeseed breeding. Thus,it is important to identify new genes that affect plant architecture and better understand the related regulatory mechanisms in rapeseed.
In this study,a new rapeseed mutant,df34,was isolated by EMS-induced mutagenesis,and it exhibited a semi-dwarf phenotype. First,the F1,F2,BC1P1and BC1P2populations were constructed to determine the genetic model of thedf34semi-dwarf trait. Then,the locus was fine-mapped using a bulked segregant analysis coupled with whole-genome sequencing (BSA-Seq) and mapbased cloning strategies (Takagiet al.2013). Finally,the multiple omics methods were combined to predict the candidate genes. This study provides a novel major QTL affecting semi-dwarf architecture and a new genetic resource for use in the breeding of semi-dwarfB.napuscultivars.
2.Materials and methods
2.1.Plant materials and population development
In this study,the semi-dwarf mutantdf34was obtained by EMS-induced mutagenesis of Ningyou 18 (NY18) with a 0.3% EMS solution (W/V,Catalog M-0880,Sigma) for 18 h (Wanget al.2020). The mutagenized seeds (M0generation) were sown in a field,and the PH mutants were characterized and bagged at the flowering stage. M1seeds were then sown into independent lines. Mutants were cultivated by raising and transplantation of the seedlings with a plant spacing of 40 cm and a row spacing of 18 cm. After years of self-pollination,the stable semidwarf mutantdf34was obtained.
An F2segregated population derived from a cross between ‘NY18’ anddf34(NY18×df34) was used for genetic analysis and BSA-Seq. The F2populations of Zhongshuang 11 (ZS11)×df34and Holly×df34were constructed for fine-mapping the locus that controls thedf34semi-dwarf phenotype. ‘NY18’ and ‘ZS11’ are typical semi-winter rapeseed cultivars from China,and Holly is a winter cultivar from Canada. All the segregated populations were planted in Nanjing,Jiangsu Province,China.
2.2.Trait measurements and genetic analysis
The agronomic traits,including PH,effective branch height,effective branch number,length of the main inflorescence,internode length,internode number,silique number per plant and seeds per silique,were measured for wild-type (WT) NY18,mutantdf34and their F1at maturity following the description of Wanget al.(2020).Each measurement was repeated three times. At maturity,three plants were randomly and independently selected from NY18,df34and their F1. The plants were used to measure biomass and harvest index. Nine welldeveloped siliques on the main inflorescence adjacent to the uppermost branch were collected,and the length and seeds per silique were determined. The differences between WT and the other two groups (F1anddf34)were analyzed using Student’st-test with Origin Software(https://www.originlab.com/).
To further investigate the PH-related genetic regulation ofdf34,six generational populations were constructed,including P1(NY18),P2(df34),(NY18×df34) F1,F2,(F1×NY18) BC1P1and (F1×df34) BC1P2(Caoet al.2013),and genetic analyses were conducted using the mixed major-gene plus polygenes inheritance model of the SEA-G4F2Software package. Using the principle of the minimum Akaike’s information criterion value,the optimal genetic model of the semi-dwarf trait was determined (Caoet al.2013).
2.3.BSA-Seq analysis
At maturity,25 WT plants (H-pool) and 25 dwarf plants(D-pool) were selected and used to construct two independent bulks for BSA-Seq from the (NY18×df34) F2population. A plant genomic DNA kit (Tiangen,Beijing,China) was used to extract the genomic DNA from fresh leaves. The two bulks were constructed by mixing equal ratios of the appropriate individual DNAs. Sequencing libraries of H-pool,D-pool and the two parents were constructed,and the Illumina HiSeq™ PE150 (Illumina,Inc;San Diego,CA,USA) platform was used to resequence the samples. To ensure the quality of the sequencing data and avoid interference with subsequent comparisons,raw data were processed using the Trimmomatic Software to obtain clean data (Bolgeret al.2014). The construction of libraries,sequences and quality controls were carried out by Novogene Bioinformatics Technology Co.Ltd.(Beijing,China).The clean re-sequence data were aligned against the rapeseed reference genome “Darmor-bzh” using BWA Software (Li and Durbin 2009;Chalhoubet al.2014). The SAMtools Software was used to remove duplicates,filter out the split alignment sequence,and screen for single nucleotide polymorphisms (SNPs) and insertion/deletions(InDels) between the two parents (Liet al.2009). The functions of variation sites were annotated using the ANNOVAR tool (Wanget al.2010). The SNP-index of the two bulks was calculated using NY18 as the reference parent,with ∆(SNP-index)=SNP-index (H-pool)-SNPindex (D-pool). The sliding windows method was used to reflect the distribution of the SNP-index of offspring on the chromosomes. Any locus having a significantly different(P<0.01) Δ(SNP-index) was considered a candidate associated with PH.
2.4.Fine mapping of the QTL controlling PH
After obtaining a primary locus by BSA-Seq,SNP markers based on the Penta-primer amplification refractory mutation system (PARMS) technology were designed to fine-map the region (Zhanget al.2019). The PHs of all the individuals in the (ZS11×df34) F2and (Holly×df34) F2segregating populations were measured at maturity. The nucleic acid sequences 200-bp upstream and downstream of the selected SNP were submitted to an online primer design tool (http://www.snpway.com/) to design the primers for the PRAMS SNP markers. The primers were synthesized by Gentides Biotech Co.,Ltd.(Wuhan,China). The effects of marker genotyping were verified on the validation group. First,a polymorphic marker was designed on both sides of the interval,and used to screen the recombinant plants of the (ZS11×df34) F2population,which contained 2 555 individuals. Then,to further narrow the interval,polymorphic markers between these two markers were designed to screen the recombinant plants.
The interval was first narrowed using the (ZS11×df34)F2population. To further confirm and fine-map the interval,markers on both sides of the narrowed interval were used to screen the (Holly×df34) F2population,which contained 2 576 individuals. Then,new polymorphic markers were designed to screen these recombinants.The final fine-mapping interval was obtained when no new polymorphic markers could be designed. The open reading frames (ORFs) and their annotation information underlying the final interval were obtained from the“Darmor-bzh” and “ZS11” reference genomes ofB.napus(Chalhoubet al.2014;Sunet al.2017).
2.5.Transcriptome sequencing and analysis
Equivalent amounts of stem tips from NY18 anddf34at the stem elongation stage were collected and used for transcriptome sequencing,with three biological replicates per material. The total RNA of each sample was extracted using an RNAprep pure Plant Kit (Tiangen,China).Samples with acceptable concentration and integrity levels were used to construct sequence libraries. RNA sequencing (RNA-seq) and quality control of raw reads were performed by Novogene Bioinformatics Technology Co.Ltd.(Beijing,China) on an Illumina HiSeq 2000 platform. The clean reads were aligned to the “Darmorbzh” genome using HISAT2 (Kimet al.2019),Fragments Per Kilobase exon Model per Million mapped reads(FPKM) was used to eliminate the influence of gene lengths and sequencing discrepancies on the expression calculations,and differential expression was assessed with the DESeq2 R package (Loveet al.2014). Subsequently,Gene Ontology (GO) enrichment analysis of DEGs was conducted using the GOseq R package (Younget al.2010),and GO terms with correctedP<0.05 were considered to be significantly enriched. KOBAS 2.0 was used to perform a Kyoto encyclopedia of genes and genomes (KEGG)enrichment pathway analysis of the DEGs (Xieet al.2011).
2.6.Phytohormone measurements
The contents of endogenous phytohormones,including auxin,cytokinin,jasmonic acid,salicylic acid,abscisic acid and GA,in stem apices of NY18 anddf34were measured using liquid chromatography-tandem mass spectrometry(LC-MS/MS),for three replicates of both NY18 anddf34.The samples used for phytohormone measurements were the same as those for RNA-seq. The 50-mg samples were ground into powder and extracted with methanol/water/formic acid (15/4/1,v/v). The extraction solution was evaporated,reconstituted in 100 µL methanol/water(8/2,v/v),filtered through a 0.22-µm PTFE membrane,and then the contents of the endogenous phytohormones were determined using LC-MS/MS. The sample processing and hormone measurements were performed by Metware Biotechnology (Wuhan,China).
3.Results
3.1.Phenotypic and genetic analyses
The PH of semi-dwarf mutantdf34was significantly shorter than NY18 during the whole growth period (Fig.1-A and B). The PH ofdf34was (112.4±9.81) cm,which was significantly less (P<0.01) than that of NY18 ((180.4±3.6) cm)at the mature stage (Table 1). The height of the first effective branch,the length of the internode,the first effective branch number and the length of the main inflorescence ofdf34were all reduced compared with NY18 at the mature stage (Fig.1-C;Table 1). The silique lengths ofdf34and F1were also reduced compared with NY18 (Fig.1-D). However,the reduction in the main inflorescence length was relatively small (approximately 17.85%). The F1plants had more branches and a greater total silique number compared with WT (Table 1).
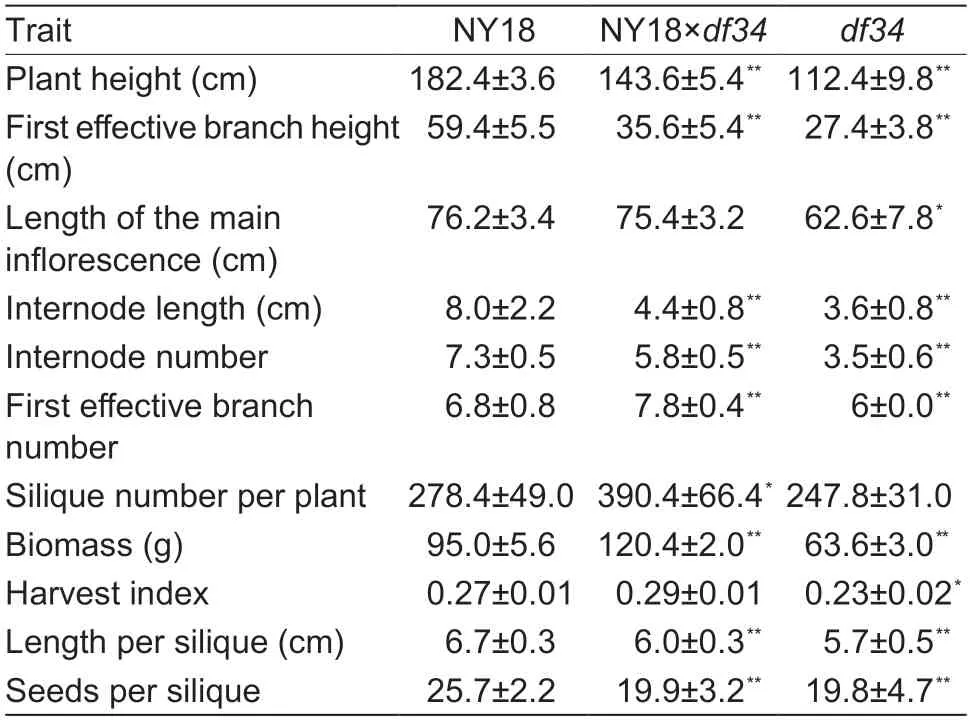
Table 1 Agronomic traits of NY18,df34 and the F1 crossed between NY18 and df34

Fig.1 The phenotypes of NY18,F1 and df34. A and B,plant heights of NY18,F1 and df34 at the bolting and mature stages,respectively. C and D,architecture of stems,branches and internodes and characteristics of siliques of NY18,F1 and df34 at the mature stage. Bars=10 cm (A and C),20 cm (B),and 2 cm (D).
The F2populations of crosses betweendf34and NY18 were used to conduct a Chi-squared test during the genetic analysis. In the (NY18×df34) F2population,which included 217 plants,there were 57,106 and 54 homozygous semi-dwarf,hybrid and WT plants,respectively,which approximately fitted the expected Mendelian inheritance ratio of 1:2:1 (χ2=0.024,P>0.05).This result indicated that the semi-dwarf trait is controlled by a semi-dominant locus. The mixed major-gene plus polygenes inheritance model was used to conduct a genetic analysis using six generational populations. 1MGAD was the best fitting genetic model,indicating that the semi-dwarf ofdf34is controlled by one major gene with additive-dominant effects (Appendix A). These results agreed with the genetic analysis.
3.2.BSA-Seq analysis
BSA-Seq was used to preliminarily map the semi-dwarf gene. The H-pool,D-pool and the two parents generated 187.7 Gb of clean bases,with Q20≥96.75% and Q30≥93.08%,indicating that the quantity of the sequence data was adequate (Appendix B). The clean data were aligned to reference genome “Darmor-bzh”,with average depths of 39.61×,25.39×,35.94× and 36.92× for NY18,df34,H-pool and D-pool,respectively. A total of 150 463 different and homozygous SNPs were identified between the two parents. After 1 000 permutation tests,the interval(17.14–43.36 Mb) of chromosome C03 was identified as the primary mapping region,at a 99% significance level,and this locus was namedBnaSD.C3(Fig.2). In this interval,the greatest SNP-index of 0.974 was with the H-pool,whereas the lowest SNP-index of 0.152 was with the D-pool. The average ∆(SNP-index) was 0.553. There were 2 785 SNPs in theBnaSD.C3region,including 123 exonic SNPs and 88 intronic SNPs (Appendix C).

Fig.2 The identification of a quantitative trait locus using a whole-genome resequencing analysis. The region between the red lines is the primary mapping region of BnaSD.C3 at a 99% significance level.
3.3.Fine mapping of BnaSD.C3
To fine-map theBnaSD.C3locus,the polymorphic PARMS SNP markers on both sides ofBnaSD.C3,i.e.,M1 (located on 24 888 384 bp of chromosome C03) and M9 (located on 32 634 525 bp),were first developed to screen the recombinant individuals of the (ZS11×df34) F2population.The primer information of PARMS SNP markers used for fine-mapping are listed in Appendix D. M1 and M9 amplified 189 and 244 recombinant individuals,respectively,among 2 555 individuals (Fig.3). Subsequently,seven other markers,M2–M8,were designed to screen the 433 recombinant individuals. The M2,M3,M4,M5,M6,M7 and M8 markers identified 91,66,61,45,30,86 and 108 recombinants,respectively. Finally,the location ofBnaSD.C3was narrowed to the interval between M4(C03: 27 515 246 bp) and M7 (C03: 29 176 115 bp) (Fig.3),corresponding to a 1.66-Mb interval in the “Darmor-bzh”genome.
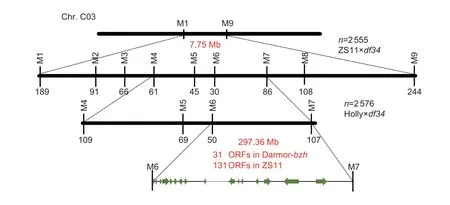
Fig.3 Map-based cloning of BnaSD.C3 using two segregated populations. Markers M1–9 were used to screen the recombinant plants in the (ZS11×df34) F2 population. Markers M4–7 were also used to screen the recombinant plants in the (Holly×df34) F2 population. The number below each marker represents the number of recombinants screened by the marker. The BnaSD.C3 interval was aligned to the “Darmor-bzh” and “ZS11” reference genomes.
To further verify and narrow theBnaSD.C3region,M4 and M7 were used to genotype the (Holly×df34) F2population at the seedling stage,which included 2 576 individuals (Fig.3). M4 and M7 identified 109 and 107 plants,respectively,as recombinants. Subsequently,these 216 recombinants were transplanted into the field to confirm their PHs,and M5 and M6 identified 69 and 50 recombinant individuals among these recombinants,respectively. After several attempts,we could not design any new SNP markers with a good genotyping effect based on PARMS technology. Finally,theBnaSD.C3region was narrowed to a 297.36-kb interval in the“Darmor-bzh” reference genome between markers M6 and M7 (Fig.3).
3.4.RNA-seq and phytohormone content analysis
In this study,20.06–25.80 million clean reads were obtained from each sample using high-throughput RNAseq. Using the criteria of false discovery rate <0.05 and|log2fold change|≥1,2 035 DEGs were identified between NY18 anddf34. Among them,the expression levels of 828 genes were up-regulated and those of 1 207 genes were down-regulated indf34(Fig.4-A). The results of GO enrichment analysis showed that some DEGs were significantly enriched in energy metabolism organs such as chloroplasts (up-regulated) and mitochondria(down-regulated),and some DEGs were involved in the biosynthesis of glycoside (down-regulated) and response to auxin (down-regulated) (Fig.4-C). This indicated that the dwarf ofdf34may be caused by the impediment of energy metabolism or auxin signal transduction. The KEGG enrichment analysis showed that the DEGs were significantly enriched in the carbohydrate metabolism(110 genes),energy metabolism (50 genes) and lipid metabolism (48 genes) pathways (Fig.4-B). Most previous dwarf mutants showed blockage of the biosynthesis or signal transduction of phytohormones(Penget al.1999;Zhaoet al.2017;Liet al.2019). In the present study,48 DEGs were enriched in signal transduction (Fig.4-B),and 36 DEGs were enriched in plant hormone signal transduction,which mainly involved the signal transduction of auxin (23 genes) and cytokinin(four genes) (Appendix E).
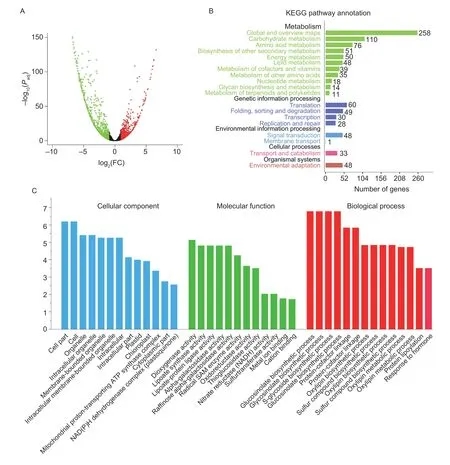
Fig.4 The results of transcriptome sequencing. A,volcano plot of differentially expressed genes. The red and green dots represent the significantly upregulated and significantly downregulated genes,respectively. B,results of the Kyoto encyclopedia of genes and genomes (KEGG) enrichment analysis of the differentially expressed genes. C,results of the Gene Ontology (GO) enrichment analysis of the differentially expressed genes.
To determine whether the semi-dwarf trait was affected by the phytohormone contents,the plant hormone levels in NY18 anddf34were measured using LC-MS/MS.Among the hormones associated with PH,the contents of trans-zeatin (a kind of cytokinin) increased significantly,but those of GA3and GA4decreased significantly indf34,and the content of auxin changed insignificantly (Appendix F). Combining the DEGs enrichment analysis and hormone content data,we speculated that the candidate genes may be related to energy metabolism and/or plant hormones,which provides a direction for finding more candidate genes in the future.
3.5.Candidate gene analysis
The fine-mapped interval was aligned to the reference genome “Darmor-bzh”,and 31 annotated or predicted genes were found underlying the 297.36-kb interval ofBnaSD.C3(Appendix G). The markers M1 and M9 were located inBnaC03g43790DandBnaC03g44090D,respectively,but did not co-separate withdf34(Fig.3).In addition to these two genes,only 10 other genes were expressed in NY18 ordf34according to the RNAseq results. However,the 10 expressed genes had no significant differences in expression level,and there were no structural variations among these genes between NY18 anddf34based on the re-sequencing results(Appendix G). In addition,the annotation information showed that the functions of these 10 genes were independent of plant architecture. Thus,no potential candidate gene forBnaSD.C3was identified.
Subsequently,theBnaSD.C3locus was aligned to reference genome “ZS11”,and the target interval corresponded to a 1.17-Mb region between SNP markers M6 and M7. The results of BSA-Seq based on the “ZS11”genome also showed that this was a candidate interval at the 99% confidence level (Appendix H). A total of 134 annotated genes were found underlying this interval in the “ZS11” reference genome. The RNA-seq analysis indicated that 66 of the 134 annotated genes were not expressed in NY18 ordf34,so these genes were excluded as candidate genes forBnaSD.C3(Appendix I). In the re-sequence results,SNPs were detected in the exons of five genes,the introns of five genes and the untranslated region of one gene (Table 2).
There was a SNP in the untranslated region ofBnaC03G0469900ZS,but the annotation information showed that this gene affects chlorophyll biosynthesis and photosynthesis (Zhouet al.2022). Among the five genes that had exonic SNPs,only the expression level ofBnaC03G0476500ZSshowed a significant change between NY18 anddf34,with the SNP causing a stopgain mutation.BnaC03G0476500ZSmainly affects plant resistance to Na+(Rausellet al.2003),but the contribution to PH variation is uncertain. The annotation information ofBnaC03G0470900ZSshowed that it is required for meiosis(Stevenset al.2004). A study of aBnaC03G0472800ZShomolog in rice showed that this gene affects root hair length,but it reportedly had no obvious effect on PH(Huanget al.2013).BnaC03G0476600ZSis involved in the regulation of chloroplasts,and its mutant is embryonic lethal (Caninoet al.2009),but its contribution to PH variation remains uncertain.BnaC03G0478900ZSencodes a tetratricopeptide repeat-like superfamily protein(Table 2),and the best-matched protein inArabidopsisis a pentatricopeptide repeat (PPR) superfamily protein(TAIR: AT1G18485.1) that can regulate mitochondrial RNA editing (Barkan and Small 2014). Mitochondria are the centers of energy metabolism in plants,providing energy for plant growth and development. For example,a mutation inBIR6encoding a PPR protein affected the assembly of mitochondrial Complex I and inhibited growth (Koprivovaet al.2010). Transcriptome analysis also showed that the DEGs were significantly enriched in mitochondria and energy metabolism terms (Fig.4).Therefore,we concluded thatBnaC03G0478900ZSwas the most likely candidate gene forBnaSD.C3. A SNP was located in the intron ofBnaC03G0466900ZS,which encodes a PHD finger family protein (Table 2). PHD finger family proteins play causal roles as regulators in gene expression. For example,OsTITANIA,a rice PHDfinger protein,causes rice dwarfism by affecting the absorption of multiple metals (Tanakaet al.2018). The gene annotation information of four other genes with SNPs in their introns indicated no obvious effects on PH.Introns play important roles in gene expression in higher plants (Xie and Wu 2002). For example,a T-DNA mutant of the 4th intron of thePFPgene ofArabidopsischanges its expression level (Yokoyamaet al.2019). Therefore,BnaC03G0466900ZSis a candidate gene forBnaSD.C3.
4.Discussion
4.1.The mutant df34 has potential application value in semi-dwarf breeding of rapeseed
Rice and wheat have attracted much scientific attention because they are major food crops,and a large number of genes have been cloned to improve their plant architecture and other agronomic traits (Wang and Li 2008;Fuet al.2022). The application of the dwarf genessd1andRht-1has significantly increased the yields of these crops and guaranteed food security (Penget al.1999;Sasakiet al.2002). The rapeseed yield may be remarkably increased by improving the plant architectureand rational close planting. Rapeseed is an important oil crop,supplying a large amount of high-quality edible oil.Since the creation and popularization of hybrid rapeseed in the 20th century,the rapeseed yield has significantly increased,but the PH also increased (Fu and Zhou 2013).The increase in PH reduces lodging resistance and is not conducive to mechanization,so it is of great significance to produce excellent dwarf germplasm resources and to clone the related major genes. Some rapeseed dwarf mutants have been obtained previously,but most of them are extreme dwarfs and susceptible to disease,with small siliques and lower seed production,which are not conducive to subsequent breeding programs (Zhaoet al.2017;Wanget al.2020).
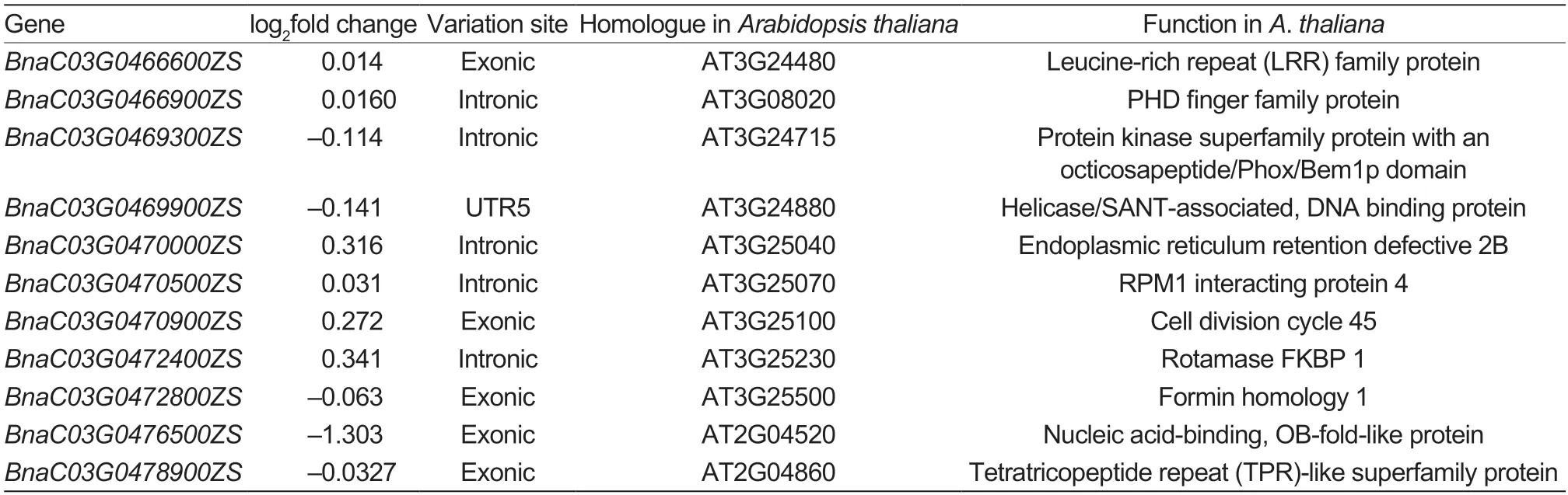
Table 2 The open reading frames obtained by aligning BnaSD.C3 to the “ZS11” reference genome,which were expressed in NY18 and df34,and the existing SNPs
In this study,a semi-dwarf mutant was obtained by EMS-induced mutagenesis. The PH of the homozygous mutantdf34was (112.4±9.81) cm (Fig.1-A and D).Compared with the WT,the F1had a super parental advantage in branching numbers (Table 1). Under the same cultivation conditions,the biomass and harvest index of (NY18×df34) F1increased,suggesting thatdf34has an important potential application value in breeding.The yields of (ZS11×df34) F1and (Holly×df34) F1are also higher than those of their parents (Appendix J). Among the reported rapeseed dwarf mutants,the branch heights,internode lengths and main inflorescence lengths were reduced dramatically compared with the WT,resulting in PH values mostly under 100 cm (Zhaoet al.2017;Yanget al.2021). In the semi-dwarf mutantdf34,the branch height and internode length were reduced by approximately half,but the main inflorescence length was reduced by approximately 17.85% (Table 2). In the semidwarf rapeseed mutantds-1,the variation in the main inflorescence length was smaller than those of the branch height and internode length (Liuet al.2010).
4.2.BnaSD.C3 is a new locus affecting rapeseed PH
The QTLs affecting PH have been obtained in all rapeseed chromosomes,but only a few major genes have been mapped and cloned. Currently,the genes responsible for PH have been detected on chromosomes A01 (Lyuet al.2022),A03 (Liet al.2019;Zhenget al.2020;Fanet al.2021;Yeet al.2022),A06 (Liuet al.2010),A08 (Liet al.2021),C04 (Yanget al.2021),C05(Zhaoet al.2019),C07 (Zhaoet al.2017) and C09(Wanget al.2020). In the present study,a candidate gene for the semi-dwarf phenotype ofdf34was mapped on chromosome C03 by map-based cloning strategies.The genesBnaMAX1andBnaTFL1on chromosome C03 also affected the PH of rapeseed (Sriboonet al.2020;Zhenget al.2020),but these two genes did not localize inBnaSD.C3. There were no other major QTLs or genes for PH cloned on chromosome C03 inB.napus,suggesting thatBnaSD.C3represents a new major locus affecting PH.
The PH of higher plants is mainly controlled by phytohormones and environment. For example,the cloned genes affecting PH in rice affect auxin,GA and BR levels (Wang and Li 2008). The genes cloned in rapeseed are also mainly associated with phytohormones (Liuet al.2010;Liet al.2019;Yanget al.2021;Lyuet al.2022). In the present study,the RNA-seq results showed that 36 DEGs were enriched in plant hormone signal transduction pathways (Appendix E),and the GA content was reduced significantly. However,the cytokinin and jasmonic acid levels increased significantly indf34(Appendix F). No genes were associated with phytohormones or PH in theBnaSD.C3locus when aligned to the “Darmor-bzh”genome. Subsequently,theBnaSD.C3locus was aligned to the “ZS11” genome,corresponding to a 1.17-Mb region that included 134 ORFs.BnaC03G0478900ZSandBnaC03G0466900ZSwere expressed in both NY18 anddf34. The annotation information showed that these two genes may be associated with PH (Table 2) (Koprivovaet al.2010). Consequently,BnaC03G0478900ZSandBnaC03G0466900ZSwere considered as the candidate genes using the multi-omics method.
4.3.New insights into the regulation of rapeseed PH
The expression levels of genes related to signal transduction change,and the GA3/GA4content is reduced significantly indf34,but there were no genes associated with phytohormones in theBnaSD.C3region. The RNA-seq results showed that 98 genes were enriched in the energy and lipid metabolic pathways (Fig.4-B).Mitochondria are the sites for energy metabolism and oil synthesis in rapeseed;therefore,mitochondrial development is of great significance. The annotation information ofBnaC03G0478900ZSshowed that the bestArabidopsisprotein match is a mitochondria-localized PPR protein. PPR proteins are involved in the RNA post-transcriptional processing of mitochondria and chloroplasts (Barkan and Small 2014). Most mutations of PPR proteins localized to mitochondria lead to delayed development,and most of the mutations localized to chloroplasts lead to etiolation and even embryonic lethality(Lee and Kang 2020). For example,BIR6encodes a mitochondrial-localized PPR protein,so thebir6mutant presented a dwarf phenotype (Koprivovaet al.2010),and mutantppr19showed a growth-defective phenotype inArabidopsis(Leeet al.2017).MTSF1encodes a new mitochondrial-targeted PPR protein,and the respiratory and photosynthetic activities were found to be altered in themtsf1mutant (Haïliet al.2013). Here,the DEGs were also involved in photosynthesis and respiratory processes(Appendix E). Therefore,we speculated that the mutation ofBnaC03G0478900ZScaused the semi-dwarfism ofdf34by affecting the development of mitochondria in rapeseed. InB.napus,intron mutations of thePFPgene encode a PHD-type zinc finger protein that changes flowering characteristics (Yokoyamaet al.2019). A PHD-type zinc finger protein has also been reported to affect the PH of rice (Tanakaet al.2018). In the present study,BnaC03G0466900ZS,which encodes a PHD-type zinc finger protein with SNPs in the intron,could not be excluded (Table 2).
PPR proteins affect fertility in rapeseed. For example,ORF2,which encodes a PPR protein,restores the fertility ofnapcytoplasmic male sterility inB.napus(Liuet al.2017),andorf224is the restorer of fertility genes in thepolCMS (Liuet al.2016). However,the regulatory mechanism by which PPR proteins affect the PH of rapeseed is not clear. Introns have important regulatory functions in gene expression (Xie and Wu 2002). The dwarf mutants identified inB.napusare almost all caused by phytohormones,not by mitochondrial development or intron mutations. The present study provides new insights into the regulation ofB.napusPH,which is of great significance for further studies on the regulatory mechanisms of PH.
5.Conclusion
In this study,a semi-dwarf rapeseed mutantdf34was obtained by EMS-induced mutagenesis and the major locusBnaSD.C3was fine-mapped to a 297.35-kb interval on chromosome C03 of the “Darmor-bzh” reference genome. There were no potential candidate genes for semi-dwarfism in this “Darmor-bzh” interval. Then,BnaSD.C3was aligned to the “ZS11” reference genome.BnaC03G0466900ZSandBnaC03G0478900ZSmay be responsible for the semi-dwarf phenotype ofdf34based on re-sequencing,RNA-seq,phytohormone analyses and gene annotation information. Thedf34mutant is an excellent germplasm resource for studying the regulation of rapeseed growth.
Acknowledgements
The work was supported by the National Natural Science Foundation of China (32172065 and 32172095),the earmarked Fund for China Agriculture Research System(CARS-12),the Central Public-interest Scientific Institution Basal Research Fund,China (Y2022QC21) and the Jiangsu Collaborative Innovation Center for Modern Crop Production,China.
Declaration of competing interest
The authors declare that they have no conflict of interest.
Appendicesassociated with this paper are available on https://doi.org/10.1016/j.jia.2023.02.017
 Journal of Integrative Agriculture2023年10期
Journal of Integrative Agriculture2023年10期
- Journal of Integrative Agriculture的其它文章
- The association between the risk of diabetes and white rice consumption in China: Existing knowledge and new research directions from the crop perspective
- Linking atmospheric emission and deposition to accumulation of soil cadmium in the Middle-Lower Yangtze Plain,China
- Genome-wide association study for numbers of vertebrae in Dezhou donkey population reveals new candidate genes
- Are vulnerable farmers more easily influenced? Heterogeneous effects of lnternet use on the adoption of integrated pest management
- lnfluences of large-scale farming on carbon emissions from cropping:Evidence from China
- Spatio-temporal variations in trends of vegetation and drought changes in relation to climate variability from 1982 to 2019 based on remote sensing data from East Asia