
Dek219 encodes the DlCER-LlKE1 protein that affects chromatin accessibility and kernel development in maize
2023-10-16 01:32:36XlESidiTlANRanZHANGJunjieLlUHanmeiLlYangpingHUYufengYUGuowuHUANGYubiLlUYinghong
Journal of Integrative Agriculture 2023年10期
XlE Si-di ,TlAN Ran ,ZHANG Jun-jie ,LlU Han-mei ,Ll Yang-ping ,HU Yu-feng ,YU Guo-wu,HUANG Yu-bi#,LlU Ying-hong
1 State Key Laboratory of Crop Gene Exploration and Utilization in Southwest China,Sichuan Agricultural University,Chengdu 611130,P.R.China
2 College of Life Science,Sichuan Agricultural University,Ya’an 625014,P.R.China
3 College of Agronomy,Sichuan Agricultural University,Chengdu 611130,P.R.China
4 Maize Research Institute,Sichuan Agricultural University,Chengdu 611130,P.R.China
Abstract Chromatin accessibility plays a vital role in gene transcriptional regulation. However,the regulatory mechanism of chromatin accessibility,as well as its role in regulating crucial gene expression and kernel development in maize (Zea mays) are poorly understood. In this study,we isolated a maize kernel mutant designated as defective kernel219(dek219),which displays opaque endosperm and embryo abortion. Dek219 encodes the DICER-LIKE1 (DCL1) protein,an essential enzyme in miRNA biogenesis. Loss of function of Dek219 results in significant reductions in the expression levels of most miRNAs and histone genes. Further research showed that the Heat shock transcription factor17 (Hsf17)-Zm00001d016571 module may be one of the factors affecting the expression of histone genes. Assay results for transposase-accessible chromatin sequencing (ATAC-seq) indicated that the chromatin accessibility of dek219 is altered compared with that of wild type (WT),which may regulate the expression of crucial genes in kernel development. By analyzing differentially expressed genes (DEGs) and differentially accessible chromatin regions (ACRs) between WT and dek219,we identified 119 candidate genes that are regulated by chromatin accessibility,including some reported to be crucial genes for kernel development. Taken together,these results suggest that Dek219 affects chromatin accessibility and the expression of crucial genes that are required for maize kernel development.
Keywords: maize,kernel development,chromatin accessibility,histone,miRNA
1.Introduction
Chromatin accessibility plays a crucial role in the regulation of gene transcription (Jiang and Pugh 2009),and it is determined by nucleosome occupancy as most transcription factors require nucleosome-deleted accessible chromatin to bind their target sequences(Minnoyeet al.2021). Nucleosomes are the basic structural units of chromatin,each nucleosome contains a histone octamer consisting of two copies of each of the four histones (H2A,H2B,H3 and H4),and a 146-bp core DNA sequence wraps around this complex (Penningset al.1994;Lugeret al.1997). Compared with the genes that have lower expression,a large degree of nucleosome deletion occurred in the promoter regions of the highly expressed genes (Leeet al.2007;Schoneset al.2008;Liet al.2014). Differences in chromatin accessibility in different tissues can affect the tissue specific expression of genes (Chen Jet al.2017),which are also closely related to cell type specificity,hotspots for phenotypeassociated genetic variants,and targets of selection during modern maize breeding (Marandet al.2021). Moreover,chromatin accessibility can also regulate the expression of C4genes and the development of the inflorescence in maize (Parvathaneniet al.2020;Sunet al.2020a;Daiet al.2022). Furthermore,the 3´ end of histone mRNA is a conserved stem-loop structure (Schümperli and Pillai 2004),the uridine-rich 7 small nuclear ribonucleoprotein(U7 snRNP),especially Sm-like protein 10 (LSM10) and LSM11 in this complex,are critical for processing the 3´ end of the histone precursor messenger RNA (premRNA;Schümperli and Pillai 2004;Sunet al.2020b),and defects in the 3´ end processing of histone mRNA result in a significant reduction in its expression (Hsinet al.2011;Gruberet al.2012;Tisdaleet al.2013).
Maize (Zea mays) is one of the most critical crops in the world. Its kernels are used as staple food and feed (Gaoet al.2022;Zhaoet al.2022). Understanding the mechanism of maize kernel development is of great significance for improving yield,and maize kernel mutants are crucial genetic materials for studying kernel development (Heet al.2019;Qiet al.2019;Zhuet al.2019). Theembryo specific(emb) mutants are one type of maize kernel mutants,which mainly show abnormal embryonic development but normal endosperm development (Clark and Sheridan 1991). They includepentatricopeptide repeat protein8522(ppr8522;Sossoet al.2012),emb-7l(Yuanet al.2019),emb12(Shenet al.2013),emb14(Liet al.2015b),emb16(Zhanget al.2013),emb8516(Magnardet al.2004) andlethal embryo1(lem1;Ma and Dooner 2004). One common characteristic of these genes is that they are all associated with plastids,which shows that a normally functioning plastid is essential for the development of the maize embryo. In addition,mutations ofSmall kernel2(Smk2;Yanget al.2017),the gene encoding glutaminase responsible for vitamin B6 biosynthesis,andBiotin1(Bio1;Suzukiet al.2020),the crucial gene responsible for biotin synthesis,also lead to defects in embryonic development.
Endosperm is the main storage tissue of maize,and it accumulates a large amount of storage proteins and starch. To date,some vital genes regulating endosperm development in maize have been discovered. For example,SUGARS WILL EVENTUALLY BE EXPORTED TRANSPORTER 4c(SWEET4c) encodes a hexose transporter that promotes the transport of nutrients,and thesweet4cmutant shows aberrant seed filling (Sossoet al.2015). Opaque2 (O2) is a basic leucine zipper(bZIP) transcription factor that plays an important role in the transcriptional regulation of maize endosperm development,and it can regulate the expression of almost allzeingenes,while theo2mutation inhibits the expression of most zein genes,resulting in the formation of opaque endosperm (Schmidtet al.1987;Liet al.2015a;Zhanet al.2018). O6/Proline responding1 (Pro1),Floury3 (Fl3),Fl4 and bZIP22 also regulate the synthesis ofzeins (Wanget al.2014a,b;Liet al.2017,2018). O2 and bZIP22 (also known as bZIP91) have also been reported to regulate starch biosynthesis (Chenet al.2016;Zhanget al.2016). Furthermore,the O2-GRAS11-EXPANSIN B-12 (EXPB12) transcriptional regulatory pathway can increase the storage capacity of endosperm cells by promoting cell expansion (Jiet al.2022),and malate dehydrogenase4 (Mdh4) affects endosperm development by regulating mitochondrial respiration,glycolysis and ATP production (Chenet al.2020).
There are few reports on the regulation of chromatin accessibility in maize kernel development. Here,we report a maize kernel mutant,dek219,which shows opaque endosperm and embryo abortion. Through positional cloning and allelic testing,we determined thatDek219encodes the DCL1 protein,a vital enzyme in miRNA biogenesis and one of the targets of selection during maize domestication (Chenet al.2022). Loss of function ofDek219inhibits the expression of most miRNAs and histone genes,and the miR167h-3p_L+1R+1-Hsf17-Zm00001d016571module may be one of the factors affecting the expression of histone genes. Furthermore,the chromatin accessibility ofdek219is altered compared with that of WT,which may regulate the expression of crucial genes in kernel development. By combining DEGs and differential ACRs between WT anddek219,we identified 119 candidate genes,including some reported crucial genes for kernel development. Therefore,Dek219affects chromatin accessibility and the expression of genes that are crucial for kernel development.
2.Materials and methods
2.1.Plant materials
The kernel mutantdek219was derived from a natural mutation of maize inbred line W22,and an ethylmethane sulfonate (EMS)-mutagenized stop-gained mutant(EMS4-0c3f3f,dek219-1) was obtained from the Maize EMS induced Mutant Database (MEMD;http://elabcaas.cn/memd/public/index.html#/;Luet al.2018). The F1heterozygous plants (dek219/+) obtained by crossing thedek219and maize inbred line B73 were self-crossed to form the F2mapping population. All materials were grown at the Chongzhou Modern Agricultural Research and Development Base,Sichuan Agricultural University.The row length was 3 m and the row width was 0.6 m,with spacing of 0.3 m between the plants within rows.Kernels (with the pericarp removed) of WT anddek219were collected at 15,20 and 25 days after pollination(DAP). Three biological replicates were collected,with each replicate sampled from the middle region of a separate well-filled and self-crossed heterozygous ear.All samples were immediately frozen in liquid nitrogen and stored at–80°C.
2.2.Measurement of kernel length,width,thickness and 100-kernel weight
Mature kernels of WT anddek219were collected from the middle region of 10 well-filled and self-crossed heterozygous ears. Kernel length,width,and thickness were examined inWT anddek219by randomly selecting 10 kernels of each ear. Next,100-kernel weight was obtained as the average weight of three repeated measurements of 100 mixed kernels from 10 ears using an electronic balance.
2.3.Measurement of starch
The total starch contents in WT anddek219kernels were measured using a starch assay kit (Megazyme,Ireland). The brief method description is as follows:First,100 mg kernel flour was weighed and 0.2 mL of 80% aqueous ethanol was added to aid dispersion;next,3 mL thermostable a-amylase was added immediately.The samples were incubated in a boiling water bath for 6 min,and then placed in a water bath at 50°C,and 0.1 mL amyloglucosidase was added. After full mixing,the samples were incubated at 50°C for 30 min. Duplicate aliquots (0.1 mL) of the diluted solution were transferred to the bottom,3 mL of glucoseoxidase/peroxidase(GOPOD) reagent was added,and the samples were incubated at 50°C for 20 min. With ddH2O as a negative control and glucose as a positive control,the absorbance of each sample was measured at 510 nm and the starch content was calculated.
2.4.Measurement of protein content
Mature kernels of WT anddek219were immersed in water to separate the endosperm from the embryo and pericarp,and the endosperm was then ground into a fine powder. A 50-mg sample of endosperm powder was weighed and added into 0.5 mL zein extraction buffer(3.75 mmol L–1sodium borate,2% β-mercaptoethanol,0.3%sodium dodecyl sulfate (SDS),70% ethanol,and pH 10)for zein extraction. In addition,0.5 mL non-zein buffer(12.5 mmol L–1sodium borate,2% β-mercaptoethanol,5%SDS,and pH 10) was used to extract non-zein protein.Three biological replicates were performed. Protein content was quantified using the BCA Protein Assay Kit(Thermo ScientificTM) in accordance with manufacturer’s protocol.
2.5.Light microscopy
WT anddek219kernels were collected at 10 and 25 DAP from the same ear and paraffin preparation was performed by cutting along the longitudinal axis. The sections were fixed in a formalin-acetic acid-alcohol mixture and dehydrated in an ethanol gradient series of 50,60,70,85,95,and 100% ethanol. After the acetone was replaced and paraffin was infiltrated,sections were embedded and cut.
2.6.Map-based cloning and allelic test
The F2segregation population was constructed by crossingdek219with B73. The bulked segregant analysis (BSA)strategy with the mixed F2segregation population was used for the preliminary screening of linkage markers. Mutant and WT samples were a mixed pool of 20 mutant and WT kernels from the F2segregation population,respectively.The F2population DNA was used for the final mapping.Based on polymorphisms between the W22 and B73 reference genomes,molecular markers for fine mapping were designed. The sequences of the primers used for mapping are listed in Appendices A and B. To confirm the candidate gene obtained by map-based cloning,we performed an allelic test. An EMS-mutagenized allelic mutant (EMS4-0c3f3f,dek219-1) ofdek219was obtained from MEMD,and the allelic test was carried out between heterozygousdek219/+anddek219-1/+.
2.7.RNA extraction
Trizol reagent (Invitrogen,Carlsbad,USA) was used to extract total RNA from each sample. The amount and purity of the total RNA were detected using a NanoDrop ND-1000 (NanoDrop,Wilmington,USA) and an Agilent 2100 with a minimum RNA integrity number (RIN)threshold value of >7.0.
2.8.miRNA analysis
A small RNA library was prepared using 1 µg of total RNA according to the protocol of the TruSeq Small RNA Sample Prep Kit (Illumina,San Diego,USA),and two small RNA libraries were constructed from equal amounts of mixed RNA from three biological replicates of WT anddek219kernel samples collected at 15 DAP. Small RNA sequencing was performed by the LC-Biotech Co.,Ltd.,Hangzhou,China. Data were processed and analyzed as previously described (Zhang Xet al.2019). Raw reads were subjected to an in-house program,ACGT101-miR (LC Sciences,Houston,Texas,USA),to remove adapter dimers,junk,low complexity reads,common RNA families (rRNA,tRNA,snRNA,and snoRNA) and repeats. Subsequently,unique sequences with lengths of 18 to 25 nucleotides were mapped to specific species precursors in miRBase 22.0 by a BLAST search to identify the known miRNAs and novel 3p-and 5p-derived miRNAs. Length variations at both 3´ and 5´ ends and one mismatch inside of the sequence were allowed in the alignment.
The unique sequences mapping to specific species of mature miRNAs in hairpin arms were identified as known miRNAs. The unique sequences mapping to the other arm of a known specific species precursor hairpin opposite to the annotated mature miRNA-containing arm were considered to be novel 5p-or 3p-derived miRNAs.The remaining sequences were mapped to other selected species precursors (with the exclusion of a specific species) in miRBase 22.0 by a BLAST search,and the mapped pre-miRNAs were further BLASTed against the specific species genomes to determine their genomic locations. We defined the above two cases as known miRNAs. The unmapped sequences were BLASTed against the specific genomes,and the hairpin RNA structures containing sequences were predicted from the flanking 120 nt sequences using RNAfold Software(http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi). miRNAs with at least a twofold change in expression (i.e.,log2fold change>1 or log2fold change<–1) and at least 10 reads in a dataset were identified here as differentially expressed miRNAs. Raw sequence data for the small RNAs in this study can be found in the National Center for Biotechnology Information Sequence Read Archive(http://www.ncbi.nlm.nih.gov/sra) under accession number SRP375987.
The expression of miR167h-3p_L+1R+1 in WT anddek219at 15,20 and 25 DAP was validated using quantitative real-time polymerase chain reaction (qRTPCR). The first-strand cDNA of this miRNA was synthesized using the miRcute miRNA First-Strand cDNA Synthesis Kit (TianGen,Beijing,China). qRT-PCR was then carried out to analyze the expression of miR167h-3p_L+1R+1 using the Bio-Rad iQ5 (Bio-Rad,USA)following the instructions for the miRcute miRNA qRTPCR Detection Kit (SYBR Green) (TianGen,Beijing,China). Three technical replicates were performed,and 5S rRNA was used as the internal reference. The qRTPCR primer is listed in Appendix C.
2.9.Transcriptome analysis
Each transcriptome sequencing library was prepared using approximately 15 µg of total RNA. A total of six transcriptome sequencing libraries were constructed using each biological replicate of the WT anddek219kernel samples collected at 15 DAP. Transcriptome sequencing was performed by the LC-Biotech Co.,Ltd.,Hangzhou,China. Data were processed and analyzed as previously described (Zhang Xet al.2019). Briefly,clean reads were obtained from the raw reads after filtering the adapter sequences and the empty,low-quality and one-copy tags. Reads were aligned to the maize reference gene set based on the B73 genome using the SOAPaligner/SOAP2 Program. Gene expression was quantified as fragments per kb per million reads (FPKM).The differentially expressed genes were selected as those with log2fold change>1 or log2fold change<–1 and with statistical significance (P-value<0.05) by an R package. Raw sequence data for the transcriptome in this study can be found in the National Center for Biotechnology Information Sequence Read Archive(http://www.ncbi.nlm.nih.gov/sra) under accession number SRP375987.
Furthermore,the expression ofDek219,Hsf17,histone H2A,H2BandH3in WT anddek219at 15,20 and 25 DAP and the expression of 22 crucial genes for kernel development in WT anddek219at 15 DAP were validated using qRT-PCR. The Fast Quant RT Kit (TianGen,Beijing,China) was used to synthesize the first strand cDNAs of these genes. qRT-PCR was then performed using the Bio-Rad iQ5 (Bio-Rad,USA) according to the SuperReal PreMix Plus (SYBR Green) instructions(TianGen,Beijing,China). All reactions were performed with three technical replicates,andglyceraldehyde-3-phosphate dehydrogenase(GAPDH) was used as the internal reference for expression. The qRT-PCR primers are listed in Appendix C.
2.10.Phylogenetic analysis
The protein sequences of maize LSM were downloaded from the Maize Genetics and Genomics Database(maizeGDB,https://www.maizegdb.org/),and the protein sequence of human LSM10 was downloaded from National Center for Biotechnology Information (NCBI,https://www.ncbi.nlm.nih.gov/). Protein sequences were aligned using MUSCLE in the MEGA5.1 Software. The neighbor-joining algorithm was used to evaluate the evolutionary distances. The bootstrap method was used to perform phylogeny testing,with 1 000 replicates.
2.11.Preparation and transfection of maize leaf protoplasts
The protoplasts of maize leaves were prepared according to Yooet al.(2007),and adapted as follows: the young leaves of maize seedlings were cut into 0.5 mm strips,which were then incubated in the enzyme solution at 25°C for 6–8 h;the protoplasts were collected by horizontal centrifugation (100×g,5 min),and resuspended in W5 solution (154 mmol L–1NaCl,125 mmol L–1CaCl2·2H2O,5 mmol L–1KCl,2 mmol L–1morpholinoethanesulfonic acid and pH to 5.8 with KOH). The protoplasts were incubated in ice for 30 min and collected by horizontal centrifugation(100×g,10 min). The protoplasts were then resuspended in MMG solution (0.4 mol L–1mannitol,4 mmol L–1morpholinoethanesulfonic acid,15 mmol L–1MgCl2·6H2O and pH to 5.6 with KOH) at a final concentration of 2×106cells mL–1. For transfection,100 µL of protoplasts were mixed with plasmid DNA (10–20 µg),and then 110 µL of freshly prepared polyethylene glycol (PEG) solution was added. The samples were incubated for 15 min at room temperature in the dark before the addition of 440 µL W5 solution. The solution was then mixed by gently inverting the tube and centrifuged at 100×g for 2 min. The protoplasts were washed once with 1 mL of W5 solution,resuspended in 500 µL of W5 solution,and cultured in darkness at 28°C for 12–18 h for subsequent experiments.
2.12.Transient expression analysis
To determine the regulatory relationships between miR167h-3p_L+1R+1 andHsf17,and betweenHsf17and the promoter ofZm00001d016571,transient expression analysis was conducted. Two precursors of maize miR167h-3p_L+1R+1,pre-miR167h1/167h2 and the coding sequences ofHsf17were cloned and inserted into the effector construct PUbi:β-Glucuronidase(GUS) by replacing theGUSreporter gene,and driven by the maize ubiquitin promoter (pUbi:miR167h1,pUbi:miR167h2 and pUbi:Hsf17). miR167h-3p_L+1R+1 target sequences ofHsf17were then cloned and inserted into the reporter construct P35S:Renilla luciferase(REN),which was located in the 3´ UTR of the reporter geneREN(P35S:REN:Hsf17).Zm00001d016571promoter fragments of different lengths were cloned into another reporter construct pPromoter:Luciferase(LUC). pUbi:GUSwas used as an internal construct. The reporter construct,the effector construct,and the internal construct were then combined at a molar ratio of 2:2:1 and co-transformed into maize leaf protoplasts. Three biological replicates were performed. The protoplasts were cultured in darkness at 28°C for 12 h,and the activities of GUS,REN and LUC were analyzed using a Luminoskan™ Ascent (Thermo,USA).
To analyze the regulatory effects of Hsf17 and Zm00001d016571 on the expression of histone genes,the coding sequence ofZm00001d016571was cloned into PUbi:GUSby replacing theGUSreporter gene(pUbi:Zm00001d016571). Next,pUbi:Hsf17(OE-Hsf17)and both pUbi:Hsf17and pUbi:Zm00001d016571(OEHsf17+Zm00001d016571) were transiently overexpressed in maize leaf protoplasts. Three biological replicates were performed. The amount of plasmid DNA and solutions used in the protoplast transfection system were scaled up five folds. Total RNA was extracted from the protoplasts,and the expression levels ofHsf17,Zm00001d016571,histone H2A,H2BandH3were analyzed using qRT-PCR.The primers used for transient expression analysis are listed in Appendix C.
2.13.Electrophoretic mobility shift assay (EMSA)
The coding sequence ofHsf17was cloned into pET32a,and recombinant His-Hsf17was purified by the Ni-NTA His Bind purification Kit (Novagen,Germany) according to the manufacturer’s instructions. An oligonucleotide probe was synthesized and labeled with biotin at the 5´end by Songon (Shanghai,China). Native-PAGE was used for electrophoresis. After electrophoresis,the binding reactions were transferred to a nylon membrane,and then the transferred DNA was crosslinked to the membrane by UV light. Biotin-labeled DNA was detected using the LightShift Chemiluminescent EMSA kit (Thermo Scientific). The probe sequence is listed in Appendix C.
2.14.ATAC sequencing
WT anddek219kernel samples taken at 15 DAP were processed according to the ATAC-Seq protocol(Buenrostroet al.2013). The numbers of cells were approximately 50 000,and the cell viability was above 80%. Chromatin was extracted and subjected to Tn5-mediated labeling and adapter incorporation according to the manufacturer’s protocol,and treated for 30 min at 37°C. A Qiagen MinElute PCR Purification Kit was used to purify the DNA. A DNA-based fluorometric assay and automated capillary electrophoresis (Agilent)were used to assess the quality of the libraries. Finally,Illumina NovaSeq 6000 sequencing was used to sequence the qualified Illumina paired-end libraries (50 bp×2,Shanghai BIOZERON Co.,Ltd.,China). The raw paired end reads were trimmed and quality controlled by Trimmomatic with parameters (SLIDINGWINDOW:4:15 MINLEN:75) (version 0.36 http://www.usadellab.org/cms/?page=trimmomatic). Then the clean reads were separately aligned to the reference genome with theorientation mode using bowtie2 (http://bowtie-bio.sourceforge.net/bowtie2/manual.shtml) Software. Modelbased analysis for ChIP-seq (MACS) was used for peak calling,and the candidate peak region was extended to be long enough for modeling. Dynamic Possion Distribution was used to calculated theP-value of the specific region based on the unique mapped reads. The region would be defined as a peak whenP-value<1E–03. Raw sequence data for ATAC sequencing in this study can be found in the National Center for Biotechnology Information Sequence Read Archive (http://www.ncbi.nlm.nih.gov/sra)under accession number SRP375987.
3.Results
3.1.The dek219 mutant displays defective embryos and endosperm
Thedek219mutant was derived from a natural mutation of W22. The mutant exhibited a stable phenotype of defective kernels,opaque endosperm,early embryonic lethality and no seedlings,in multiple environments(Fig.1-B–D). The F2kernels ofdek219exhibited 3:1 segregation (354:121,χ2=0.057<χ20.05=3.84),indicating that the defective kernel phenotype is controlled by a recessive mutation. At maturity,the 100-kernel weight of thedek219homozygous mutant was 13.26 g,or approximately 40% lower than that of the WT (Fig.1-E).The kernel length,width and thickness of thedek219homozygous mutant were 9.67,6.6 and 3.61 mm,respectively,which were also significantly reduced compared with the those of the WT (Fig.1-F–H). These results indicated thatdek219has defects in embryonic and endosperm development. Furthermore,an analysis of the starch contents of mature kernels showed that the total starch content indek219was significantly higher than that in WT (Fig.1-I),and the contents of total proteins and zeins indek219were significantly lower than those in WT(Fig.1-J).

Fig.1 Phenotypic analyses of dek219 mutants. A,a self-pollinated ear of the F1 heterozygous plants (dek219/+),where arrows indicate defective kernels. Scale bar,1 cm. B, randomly selected mature wild type (WT) (top) and dek219 (bottom) kernels. The embryo of dek219 exhibited developmental abnormalities. Scale bar,1 cm. C, transverse sections of WT (left) and dek219 (right)mature kernels. dek219 kernels showed opaque endosperm and the absence of an embryo. Scale bar,1 cm. D, longitudinal sections of WT (left) and dek219 (right) mature kernels. dek219 kernels showed opaque endosperm and the absence of an embryo. Scale bar,1 cm. E–J,100-kernel weights, kernel lengths, kernel widths, starch contents,kernel thicknesses, and protein contents of the WT and dek219 mature kernels. The early apical-basal axis of dek219 embryos was not established,and embryonic development had been arrested before the early transition stage. Bars mean SD (n=3). * and **,significant at P<0.05 and P<0.01,respectively by the Student’s t-test. K–N, paraffin sections of 10 days after pollination (DAP) (K and L) and 25 DAP (M and N) WT (K and M)and dek219 (L and N) kernels. Scale bar,500 µm.
To analyze the kernel development ofdek219,we sectioned and compared WT and mutant kernels from the same ears at 10 and 25 DAP. At 10 DAP,the apical-basal axis of the WT embryo had been established and was in the transition stage from the proembryo to the coleoptilar stage embryo (Fig.1-K),whereas thedek219embryo was not yet visible (Fig.1-L). At 25 DAP,the WT embryo had advanced to the maturation stage,and the scutellum of the WT embryo was separated from the embryo axis and had distinct shoot apical and root meristems (Fig.1-M). In contrast,the early apical-basal axis of thedek219embryo was not established,and the embryonic development had been arrested before the early transition stage(Fig.1-N). At 25 DAP,the starchy endosperm (SE) cells of the WT anddek219had developed normally,but the SE cells of the WT were more filled than those ofdek219(Appendix D-a and b). Moreover,the ingrowths of the WT anddek219developed normally in the basal endosperm transfer layer (BETL) cells,but the ingrowths of the WT were more dense (Appendix D-c and d).
3.2.Positional cloning and allelic testing confirmed that Dek219 encodes DCL1 protein
An F2segregation population derived from the selfpollinated F1(B73×dek219/+) plants was used for mapbased cloning. The DNA of 20 WT and mutant kernels were extracted from the F2population for BSA. Using 94 pairs of molecular markers distributed on 10 chromosomes(Appendix A),we found that the mutation was linked to the marker umc1354 on chromosome 1. Using 96 mutant individuals,dek219was roughly mapped to a region of approximately 3 Mb between markers Ind1-2.2 and Ind1-5.2. The mutant was fine-mapped using an additional 900 mutants and more molecular markers,which further narrowed the range to a 120-kb physical region between markers Ind4.2 and Ind4.8 (Appendix B). This region contains six annotated genes (Fig.2-A). These six genes were amplified using PCR and sequenced,and the sequencing analysis revealed that a T-base is inserted into exon 12 ofZm00001d027412,which causes a frameshift mutation,followed by a premature stop codon(Fig.2-B). Gene conserved domain analysis showed that the mutation resulted in the deletion of two essential RNaseIII domains and two RNA-binding domains in Zm00001d027412,which greatly affected the function of the protein (Fig.2-C). This gene encodes the DCL1 protein,which is a vital enzyme in miRNA biogenesis.
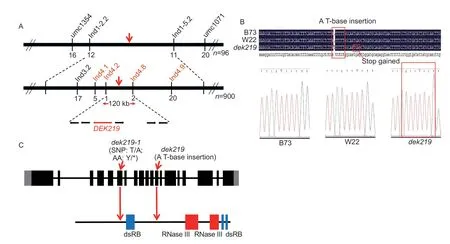
Fig.2 Map-based cloning of dek219. A, the dek219 locus was mapped to a 120-kb region on chromosome 1. The number under each molecular marker indicates the number of recombinants found in the population. B, sequencing analysis of Zm00001d027412 of B73,W22,and dek219. dek219 has a T-base insertion in exon 12,which causes a frameshift mutation,followed by a premature stop codon. C,schematic diagram of the Dek219 gene with indications of the mutation sites (top) and protein conserved domains(bottom). Black boxes represent coding regions;gray boxes represent the 5´ and 3´ untranslated regions;and lines represent introns. SNP,single-nucleotide polymorphism;AA,amino acids;dsRB,double-stranded RNA binding domains.
To confirm the candidate geneZm00001d027412,we performed an allelic test. An EMS-mutagenized mutantdek219-1(stock EMS4-0c3f3f) of this gene was obtained from MEMD (http://elabcaas.cn/memd/public/index.html#/;Luet al.2018). The mutation ofdek219-1is a T-to-A substitution in exon 6,which results in the transformation of a tyrosine residue to a premature stop codon,resulting in the deletion of two RNaseIII domains and three RNA-binding domains in Zm00001d027412(Fig.2-C). Ears from self-pollinated heterozygousdek219-1exhibited segregated defective kernels at a proportion of about 25% (200:72,χ2=0.314<χ20.05=3.84),and the defective phenotype of these kernels was similar tothat of thedek219kernels(Fig.3-A–C). The allelic test between heterozygousdek219/+anddek219-1/+generated ears exhibiting 3:1 segregation (131:48,χ2=0.315<χ20.05=3.84),which confirmed thatdek219-1is allelic todek219(Fig.3-D–F). These results confirmed thatZm00001d027412is the causative gene fordek219.This gene is one of the targets of selection during maize domestication (Chenet al.2022). Moreover,Dek219was expressed throughout the cell (Appendix E-a) and in almost all tissues (Appendix E-b).
3.3.Expression levels of most miRNAs are reduced in the dek219 mutant
Dek219encodes the DCL1 protein,which is a vital enzyme in miRNA biogenesis,so a loss-of-function ofDek219is bound to affect the biosynthesis of miRNA. To analyze miRNA expression and identify the vital miRNAs that regulate maize kernel development,miRNA sequencing was performed on WT anddek219kernels (with the pericarp removed) at 15 DAP. A total of 51 differentially expressed miRNAs were identified between WT anddek219. Among them,49 miRNAs were down-regulated indek219compared with WT,and only two miRNAs were up-regulated indek219(Appendices F and G).The expression levels of most miRNA indek219were significantly reduced,indicating that the biosynthesis of miRNA was greatly inhibited in thedek219. Thefuzzytassel(fzt) mutant is a weakly allelic mutant ofdek219.The mutation offztis a G-to-A substitution in exon 17,which results in the transformation of a serine residue to an asparagine residue in the RNaseIII domain.fztonly causes a partial loss of DCL1 protein function,which affects the inflorescence development and vegetative growth of maize but has little effect on kernel development (Thompsonet al.2014),suggesting that the essential miRNAs for maize kernel development are either not affected or less affected infzt. To further identify the miRNAs that regulate maize kernel development,we compared the miRNAs that were significantly down-regulated indek219andfzt. Among these down-regulated expressed miRNAs,28 miRNAs were down-regulated only indek219,but were unaffected infzt,so they may play a crucial role in maize kernel development (Appendices H and I).
3.4.Transcriptome profiling of the dek219 mutant
To further identify the miRNA target genes and crucial genes in maize kernel development,transcriptome analysis was performed on WT anddek219kernels(with the pericarp removed) at 15 DAP. A total of 7 613 DEGs were identified between the WT anddek219.Of which,2 836 genes were up-regulated and 4 777 genes were down-regulated indek219compared with the WT. There was no significant difference in the expression ofDek219between the WT and the mutant. GO term enrichment analysis suggested that the DEGs were significantly enriched in nucleosome assembly (GO: 0006334,P-value=8.0E–17),response to heat (GO: 0009408,P-value=1.1E–14),nucleosome(GO: 0000786,P-value=7.3E–12),and other functions(Fig.4-A),indicating thatDek219may play a crucial role in regulating the nucleosome assembly and heat stress response pathways. Histones are the main components of nucleosomes,and further analysis showed that the expression of most of the histone genes were significantly down-regulated indek219(Fig.4-B;Appendix J),which may lead to the inhibition of nucleosome assembly indek219. This may affect chromatin accessibility and gene expression,and may be the cause of the defective kernel development indek219.
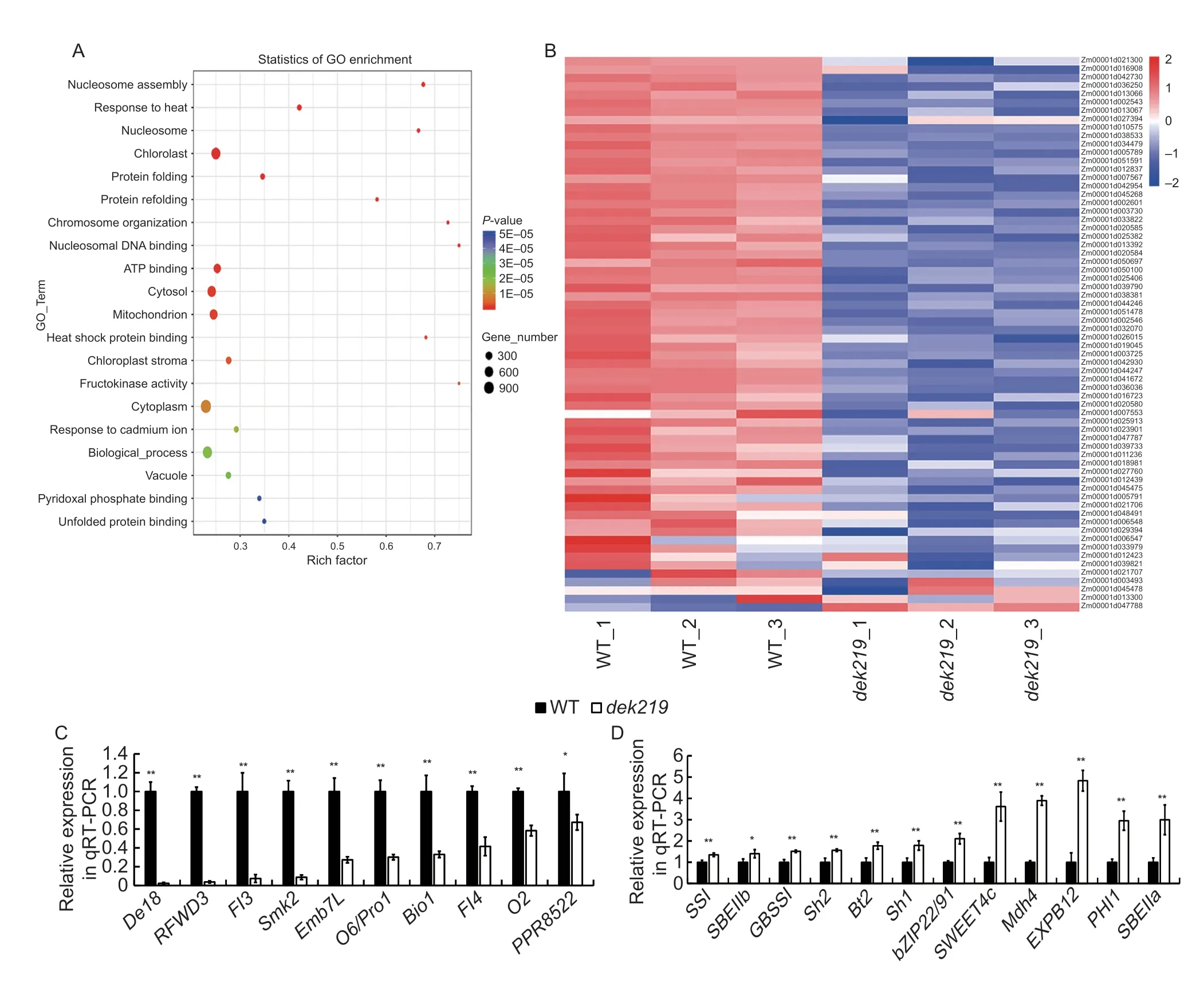
Fig.4 Transcriptome profiling of wild type (WT) and dek219. A,Gene Ontology terms of differentially expressed genes between WT and dek219. B,heat map depicting the expression levels of histone genes in WT and dek219. The expression levels of most of the histone genes were significantly down-regulated in dek219. C, expression levels of the genes involved in kernel development that were down-regulated in dek219. D, expression levels of the genes involved in kernel development that were up-regulated in dek219. Bars mean SD (n=3). * and **,significant at P<0.05 and P<0.01,respectively by the Student’s t-test.
Many crucial genes for kernel development were also significantly differentially expressed between the WT anddek219,with theembgenesEmb7L,Smk2andBio1as well as the opaque endosperm genesO2,O6andFl3all being down-regulated indek219. However,the starch biosynthesis genesBrittle2(Bt2),Shrunken2(Sh2) andGranule-bound starch synthase I(GBSSI)and the genes essential for endosperm developmentSWEET4C,Mdh4andEXPB12were all up-regulated indek219(Fig.4-C and D). The differential expression of these genes may be the direct cause of the defective kernel development indek219. However,these genes are not the direct target genes of the miRNAs,but chromatin accessibility has the potential to regulate the expression of these genes simultaneously. In addition,the expression levels of some genes in the WT anddek219at 15,20 and 25 DAP were also verified using qRT-PCR,and the results were consistent with the transcriptome data (Appendix K).
3.5.The Hsf17-Zm00001d016571 module may regulate the expression of histone genes
The expression of histone genes indek219should be regulated by miRNA-target gene modules. In this study,28 miRNAs that may regulate kernel development were identified (Appendices H and I). Using the miRNA-target gene prediction tool psRNATarget (https://www.zhaolab.org/psRNATarget/,expectation≤4),we found that 184 of the 2 836 up-regulated DEGs may be the target genes of these 28 miRNAs (Appendix L). Of these 184 genes,12 were up-regulated by more than 100-fold,of which,onlyHsf17(Zm00001d033987) had an FPKM value higher than 100 indek219,with an average of 186.63. However,the highest average FPKM value of the other 11 genes was 13.98.Hsf17is a heat shock transcription factor whose expression was up-regulated by 181-fold (Appendix M). Furthermore,Hsf17was expressed at extremely low levels in almost all tissues in the normal environment(Appendix N-a). Therefore,Hsf17was hypothesized to be one of the candidate genes for the regulation of histone gene expression.
Defects in the 3´ end processing of histone mRNA resulted in a significant reduction in its expression (Hsinet al.2011;Gruberet al.2012;Tisdaleet al.2013),U7 snRNP is critical for processing the 3´ end of histone pre-mRNA (Sunet al.2020b),and U7 snRNP complex proteins LSM10 and LSM11 play an essential role in this process (Schümperli and Pillai 2004;Sunet al.2020b).To determine whetherHsf17regulates the expression of histone genes,and whether this process is completedviaregulating the function of U7 snRNP,we further investigated one of the vital proteins,LSM10,in this study. There is no gene annotated asLSM10in maize,so we conducted a phylogenetic analysis of 12 maizeLSMgenes and humanLSM10.The results revealed two genes clustered into a clade with humanLSM10,which may have functions similar to those of humanLSM10(Fig.5-A). The expression ofZm00001d025239showed no significant difference between the WT anddek219.However,the expression ofZm00001d016571was significantly down-regulated indek219,and this gene was almost unexpressed indek219(Fig.5-B). Furthermore,Zm00001d016571was mainly expressed in kernels(Appendix N-b),whileZm00001d025239was expressed in all tissues (Appendix N-c). These results suggested thatZm00001d016571plays an essential role in kernel development.

Fig.5 The Hsf17-Zm00001d016571 module may regulate the expression of histone genes. A, phylogenetic tree of 12 maize LSM genes and human LSM10. The neighbor-joining algorithm was used to evaluate the evolutionary distances. The numbers at the nodes represent the percentage of 1 000 bootstraps. The scale bar indicates the average number of amino acid substitutions per site. B, expression of Zm00001d016571 and Zm00001d025239 in wild type (WT) and dek219. C, Hsf17 significantly inhibited Zm00001d016571 promoter activity. D,electrophoretic mobility shift assay of the binding of Hsf17-His to the TTCTAG motif.E,expression of Hsf17 in the control,OE-Hsf17,and OE-Hsf17+Zm00001d016571. F, expression of Zm00001d016571 in the control and OE-Hsf17+Zm00001d016571. G, expression of Zm00001d016571 in the control and OE-Hsf17. H, expression of histone_H2A in the control,OE-Hsf17,and OE-Hsf17+Zm00001d016571. I,expression of histone_H2B in the control,OE-Hsf17,and OE-Hsf17+Zm00001d016571. J,expression of histone_H3 in the control,OE-Hsf17,and OE-Hsf17+Zm00001d016571.LUC,luciferase;GUS,glucuronidase. OE-Hsf17 represents transient overexpression of Hsf17 in maize leaf protoplasts;OEHsf17+Zm00001d016571 represents simultaneous transient overexpression of Hsf17 and Zm00001d016571 in maize leaf protoplasts. Bars mean SD (n=3). **,significant at P<0.01 by the Student’s t-test.
To verify whetherZm00001d016571is regulated by Hsf17,transient expression analysis of theZm00001d016571promoter fragments of different lengths and Hsf17 was performed. Hsf17 did not significantly regulate the 156 bp promoter fragment upstream from the translation start codon,but it significantly inhibited the 234,292,1 440 and 2 659 bp promoter fragments upstream from the translation start codon (Fig.5-C).These results indicated that Hsf17 inhibits the expression ofZm00001d016571,and the 156 to 234 bp fragment upstream of the start codon (i.e.,the–156 to–234 bp fragment) in theZm00001d016571promoter is most likely to contain Hsf17 binding motifs. Therefore,we predicted that the–156 to–234 bp fragment contains an Hsf transcription factor binding motif,TTCTAG,using the plant promoter analysis site Plantpan3.0 (http://plantpan.itps.ncku.edu.tw/). EMSA further verified that Hsf17 can bind to the TTCTAG motif (Fig.5-D),indicating that Hsf17 can inhibit the expression ofZm00001d016571by binding to this motif.
We analyzed the regulatory effects of Hsf17 and Zm00001d016571 on the expression of histone genes by transient overexpression ofHsf17(OE-Hsf17) and simultaneous transient overexpression ofHsf17andZm00001d016571(OE-Hsf17+Zm00001d016571) in protoplasts. Compared with the control,the expression levels ofHsf17were significantly up-regulated in OEHsf17and OE-Hsf17+Zm00001d016571(Fig.5-E),and the expression level ofZm00001d016571was significantly up-regulated in OE-Hsf17+Zm00001d016571(Fig.5-F),but significantly down-regulated in OE-Hsf17(Fig.5-G),indicating thatHsf17andZm00001d016571were overexpressed in the protoplasts,and that Hsf17 significantly inhibited the expression ofZm00001d016571.Moreover,compared with the control,the expression levels ofhistone H2A,H2BandH3were all downregulated in OE-Hsf17(Fig.5-H–J). However,compared with OE-Hsf17,the expression levels of these histone genes in OE-Hsf17+Zm00001d016571were recovered and significantly up-regulated (Fig.5-H–J). These results suggested that the expression of some histone genes is significantly inhibited by Hsf17,and this regulation is completed by Zm00001d016571. Taken together,our results indicated that theHsf17-Zm00001d016571module may regulate the expression of the histone genes.
3.6.Dek219 affects chromatin accessibility,which may regulate gene expression and kernel development
Chromatin accessibility in the WT anddek219was analyzed using ATAC-seq. The abundance of reads obtained by sequencing in the gene region of the WT was significantly lower than that ofdek219,which indicated thatdek219has higher chromatin accessibility than the WT (Fig.6-A). Using MACS2 (Zhanget al.2008),7 128 and 7271 ACRs were detected in the sequencing results of WT anddek219,respectively (Appendices O and P). We further identified the differential ACRs between WT anddek219,and the genes associated with these differential ACRs. The results revealed a total of 308 genes with differential ACRs (Appendix Q). Moreover,92 of the 308 genes annotated by the GO database (http://bioinfo.cau.edu.cn/agriGO/) corresponded to eight major GO terms: regulation of post-embryonic development(GO: 0048580,P-value=0.013),anatomical structure formation involved in morphogenesis (GO: 0048646,P-value=0.023),molecular function (GO: 0003674,P-value=0.023),molecular transducer activity (GO:0060089,P-value=0.024),transferase activity,transferring glycosyl groups (GO: 0016757,P-value=0.032),transferase activity,transferring hexosyl groups (GO:0016758,P-value=0.044),regulation of multicellular organismal process (GO: 0051239,P-value=0.044),and response to temperature stimulus (GO: 0009266,P-value=0.045) (Fig.6-B),which may play a crucial role in plant development. By analyzing the DEGs and differential ACRs in this study,we found among these 308 genes,69 were down-regulated and 50 were up-regulated indek219(Appendix Q). These 119 genes may play a regulatory role in maize kernel development and are the candidate genes for maize kernel development regulation by chromatin accessibility. Furthermore,some previously reported vital genes for kernel development are included among these 119 genes,such as theembgenesSmk2andPPR8522,the opaque endosperm genesO2andO6,and thestarch biosynthesis genesBt2andStarch branching enzymeIIa(SBEIIa) (Fig.6-C;Appendix Q). Of which,Smk2,PPR8522,O2andO6were significantly down-regulated indek219,whereasBt2andSBEIIawere significantly up-regulated indek219(Fig.4-C and D). These genes all have differential ACRs between WT anddek219in their promoters (Fig.6-C). Therefore,the differences in their expression may be caused by the regulation of chromatin accessibility,which thus affect kernel development. Moreover,the exact functions of most of the 119 candidate genes with differential ACRs have not been reported previously. For instance,Zm00001d029563encodes a PPR protein that may play a crucial role in kernel development,and its expression was significantly down-regulated indek219,with differential ACRs in the promoter. In addition,Zm00001d044086,which encodes a single stranded DNA binding protein,exhibited a trend similar to that ofZm00001d029563(Fig.6-C;Appendix Q). The expression of these two genes may be influenced by chromatin accessibility and involved in the regulation of kernel development.
4.Discussion
4.1.Dek219 participates in a novel regulatory pathway that is essential for maize kernel development
The loss of function ofDek219resulted in small and defective kernels,opaque endosperm and embryonic mortality (Fig.1-B–D).Dek219encodes the DCL1 protein,a vital enzyme in miRNA biogenesis. We also conducted phylogenetic analysis of the DCL proteins in maize andArabidopsis,and Dek219 (Zm00001d027412)showed high similarity withArabidopsisDCL1 but low similarity to the other DCL proteins of maize andArabidopsis(Appendix E-c). This is consistent with previous reports (Qianet al.2011;Zhang Zet al.2019). The main function ofArabidopsisDCL1 is to produce mature miRNAs (Parket al.2002),which further indicated that Dek219 plays a crucial role in the biogenesis of miRNA. In addition,theArabidopsisdcl1mutant is also embryonic lethal (Schaueret al.2002),and strongDCL1RNA interference knockdown also results in developmental arrest in rice (Liuet al.2005).These findings indicate that DCL1 is essential for kernel development,and its function is conserved,although the specific mechanism by which DCL1 regulates plant kernel development has not been reported so far. In this study,we found that maize DCL1 regulates kernel development by affecting chromatin accessibility,which represents a novel regulatory pathway different from all known maize kernel development mechanisms (Daiet al.2021). Furthermore,we performed candidate gene association analysis forDek219using the data obtained from MaizeGo (http://www.maizego.org/Resources.html;Yanget al.2014;Liuet al.2017).SNP4473 (single nucleotide polymorphism),SNP5832,SNP6165,SNP6186,SNP6654 and SNP7143 inDek219were found to be significantly associated with kernel length (Appendix R),and these SNPs were in strong linkage disequilibrium (LD,r2>0.9). A total of 500 maize inbred lines were classified into two haplotype groups based on these six SNPs,and the inbred lines with Haplotype2 (Hap2) exhibited significantly longer kernels than those with Hap1. These six SNPs are all located at the coding region,however,only SNP4473 results in an amino acid residue substitution from glycine to aspartic acid,which may be responsible for regulating kernel length. Moreover,this gene is one of the targets of selection during maize domestication(Chenet al.2022),and SNP4473 is located in the selected region,so it has potential application value for breeding.
4.2.Dek219 may regulate the expression of histone genes via the miR167h-3p_L+1R+1-Hsf17-Zm00001d016571 module
Through a comparative analysis withfzt(Thompsonet al.2014),we identified 28 candidate miRNAs that regulate kernel development in this study. miR164e plays a vital role in maize kernel development,and overexpression of zma-miR164e results in the failure of seed formation inArabidopsis(Liuet al.2020). The expression of miR164e was significantly down-regulated indek219,although this may not be the cause of the defective kernels ofdek219.
Histones are the main components of nucleosomes(Penningset al.1994;Lugeret al.1997),and nucleosome occupancy determines chromatin accessibility,which plays a critical role in gene transcriptional regulation (Jiang and Pugh 2009;Minnoyeet al.2021). Downregulation of histone gene expression may result in the inhibition of nucleosome assembly,affecting chromatin accessibility and gene expression,which may be one of the causes of defective kernel development indek219. InArabidopsis,heat stress (HS) affects chromatin accessibility and regulates gene transcription (Kumar and Wigge 2010;Pecinkaet al.2010). After HS,plants can produce HS memory to adapt to the environment (Charnget al.2006,2007;Meiri and Breiman 2009;Stiefet al.2014).Chromatin accessibility changes induced by HS are one of the bases of HS memory. FORGETTER1 (FGT1)and chromatin remodelers play crucial roles in regulating chromatin accessibility and HS memory (Brzezinkaet al.2016). Furthermore,HSFA2 and HSFA3 also play a vital role in regulating HS memory inArabidopsis(Charnget al.2007;Friedrichet al.2021),suggesting that heat shock transcription factors may be essential in the regulation of chromatin accessibility. In this study,of the 184 miRNA target genes,onlyHsf17is a heat shock transcription factor,and it was predicted to be a target of miR167h-3p_L+1R+1. This miRNA was almost unexpressed indek219(Appendices I,L and M),and qRT-PCR also confirmed the result (Appendix K). Therefore,the miR167h-3p_L+1R+1-Hsf17module was hypothesized to be one of the candidate pathways for the regulation of histone gene expression.
miR167h-3p_L+1R+1 is significantly down-regulated indek219,and its predicted target geneHsf17is significantly up-regulated indek219(Appendix S-a). To verify the regulatory effect of miR167h-3p_L+1R+1 onHsf17,transient expression analysis of miR167h-3p_L+1R+1 and the predicted target sequences ofHsf17was performed in maize leaf protoplasts. miR167h1 and miR167h2,two precursors of maize miR167h-3p_L+1R+1,significantly inhibited the activity of the reporter with target sequences ofHsf17after transient expression in protoplasts (Appendix S-b),indicating thatHsf17is the target gene of miR167h-3p_L+1R+1.
We further identifiedZm00001d016571in maize and it may have a similar function asLSM10,which plays an essential role in the 3´ end processing of histone premRNA (Schümperli and Pillai 2004;Sunet al.2020b).Defective 3´ end processing of histone mRNA results in a significant reduction in its expression (Hsinet al.2011;Gruberet al.2012;Tisdaleet al.2013). The expression ofZm00001d016571is regulated byHsf17,and Zm00001d016571 can regulate the expression of histone genes (Fig.5-B–J). Furthermore,the susceptibility of maize kernels to drought stress leads to embryo abortion(Kakumanuet al.2012). Interestingly,under drought stress,the expression levels ofZm00001d016571and most histone genes were significantly down-regulated in the ovaries of B73 at 1 DAP compared with those in the control (Appendix S-c;Kakumanuet al.2012). This finding is consistent with the expression patterns of these genes indek219,suggesting that these genes are critical in maize kernel development.
In addition,the results of candidate gene association analysis based on MaizeGo data (http://www.maizego.org/Resources.html) also showed that natural variations inDek219,Hsf17andZm00001d016571are significantly associated with the expression of multiple histone genes,and natural variations inDek219are significantly associated with the expression ofHsf17(Appendix T). These results indicated thatDek219,Hsf17andZm00001d016571could regulate the expression of histone genes,whileDek219could regulate the expression ofHsf17,which verified the transcriptional regulatory pathway ofDek219on histone gene expression found in this study. Meanwhile,Dek219,Hsf17,Zm00001d016571and the histone genes have multiple natural variations that are significantly associated with kernel-related traits (Appendix U). These results demonstrated that these genes could regulate maize kernel development.Furthermore,likeDek219,Hsf17is also one of the targets of selection during maize domestication (Chenet al.2022),and natural variations significantly associated with kernel-related traits and the expression of histone genes inDek219andHsf17are located in the selected regions,indicating that the regulation of kernel development and the expression of histone genes by these two genes may be under selection.
4.3.Differential expression of crucial genes regulated by chromatin accessibility is the direct cause of the defective kernel development
Many genes reported to be crucial for kernel development were significantly differentially expressed between WT anddek219(Fig.4-C and D). However,these genes are not the direct target genes of miRNAs,but chromatin accessibility has the ability to regulate the expression of these genes simultaneously. Thus,the differential expression of these genes,which is the direct cause of defective kernel development indek219,may be regulated by chromatin accessibility. Using ATAC-seq,we found that some of these genes have differential ACRs between WT anddek219in their promoters (Fig.6-C;Appendices O,P,Q and V),which further suggested that these genes may be regulated by chromatin accessibility. Of these genes,the expression ofSmk2,PPR8522andBio1were significantly down-regulated indek219. The loss of function of these genes results in embryonic development defects similar to those fordek219(Sossoet al.2012;Yanget al.2017;Suzukiet al.2020). The expression ofO2,O6,Fl3andFl4were also significantly down-regulated indek219,and mutations in these genes result in opaque endosperm(Schmidtet al.1987;Wanget al.2014a,b;Liet al.2015a,2017;Zhanet al.2018). The significantly down-regulated expression of these genes may be regulated by chromatin accessibility,which is the direct cause of embryo abortion and opaque endosperm indek219. Furthermore,the differential ACR (a 165 to 449 bp fragment upstream of the start codon) in theSmk2promoter was analyzed using the plant ChIP-seq database (PCBase;http://pcbase.itps.ncku.edu.tw). This fragment contains multiple motifs that may bind to both histones and transcription factors,such as the TTTGG,CCACCA and CCAAA motifs (Appendix W). Of these motifs,the CCACCA motif may bind to both histone H3 and MADS29,a regulator of early seed development in rice (Yin and Xue 2012). Changes in chromatin accessibility indek219may facilitate the binding of transcription factors to these motifs and regulate gene expression.
Except for the opaque endosperm,dek219endosperms almost developed normally (Appendix D-a and b),which may be related to the significantly up-regulated expression of some critical genes in endosperm development(Fig.4-D). Some of these genes also showed differential ACRs in their promoters (Fig.6-C;Appendix O,P,Q and V),such as the endosperm starch biosynthesis related genesBt2,Sh2,GBSSI,SBEIIaandbZIP91(Shureet al.1983;Bhaveet al.1990;Preisset al.1990;Zeemanet al.2010;Chenet al.2016),as well as the genes essential for endosperm developmentSWEET4C,Mdh4andEXPB12(Sossoet al.2015;Chenet al.2020;Jiet al.2022). The expression of these genes may be regulated by chromatin accessibility. We also analyzed the differential ACR (a 257 to 928 bp fragment upstream of the start codon)in theBt2promoter using the PCBase. This fragment also contains multiple motifs that may bind to both histones and transcription factors (Appendix X). bZIP91 can regulate the expression ofBt2by binding to the ACTCAT motif (Chenet al.2016),whereas a similar motif,ACTCATA,may bind to histone H3 (Appendix X). These results suggested that the upregulation ofBt2expression indek219may be mediated through the regulation of chromatin accessibility.
Moreover,most of the DEGs with differential ACRs in this study have not been reported previously,includingZm00001d029563andZm00001d044086(Fig.6-C;Appendices O,P and Q).Zm00001d029563encodes a PPR protein,and most PPR genes affect plant development by regulating mitochondrial or plastid functions (Sossoet al.2012;Chenet al.2017,2023;Sunet al.2018;Yuanet al.2019).Zm00001d044086encodes a single stranded DNA binding protein. Candidate gene association analysis showed that natural variations inZm00001d029563andZm00001d044086are significantly associated with kernel length,width and 100-kernel weight (Appendix Y). Therefore,these two genes are highly likely to be involved in kernel development.
5. Conclusion
We clonedDek219,which encodes the DCL1 protein,an essential enzyme in miRNA biogenesis. The loss of function ofDek219inhibited the expression of most miRNAs and histone genes. Furthermore,the miR167h-3p_L+1R+1-Hsf17-Zm00001d016571module may be one of the factors affecting the expression of histone genes.ATAC-seq indicated thatDek219affects chromatin accessibility,which may regulate the expression of crucial genes in kernel development. By analyzing the DEGs and differential ACRs between the WT anddek219,we identified 119 candidate genes that are regulated by chromatin accessibility,including some previously reported crucial genes for kernel development (Fig.7).This is a novel and vital pathway for the regulation of maize kernel development. Furthermore,bothDek219andHsf17are the targets of selection during maize domestication (Chen et al.2022),and natural variations inDek219andHsf17are significantly associated with maize kernel related traits and the expression of histone genes,with potential application value for breeding.
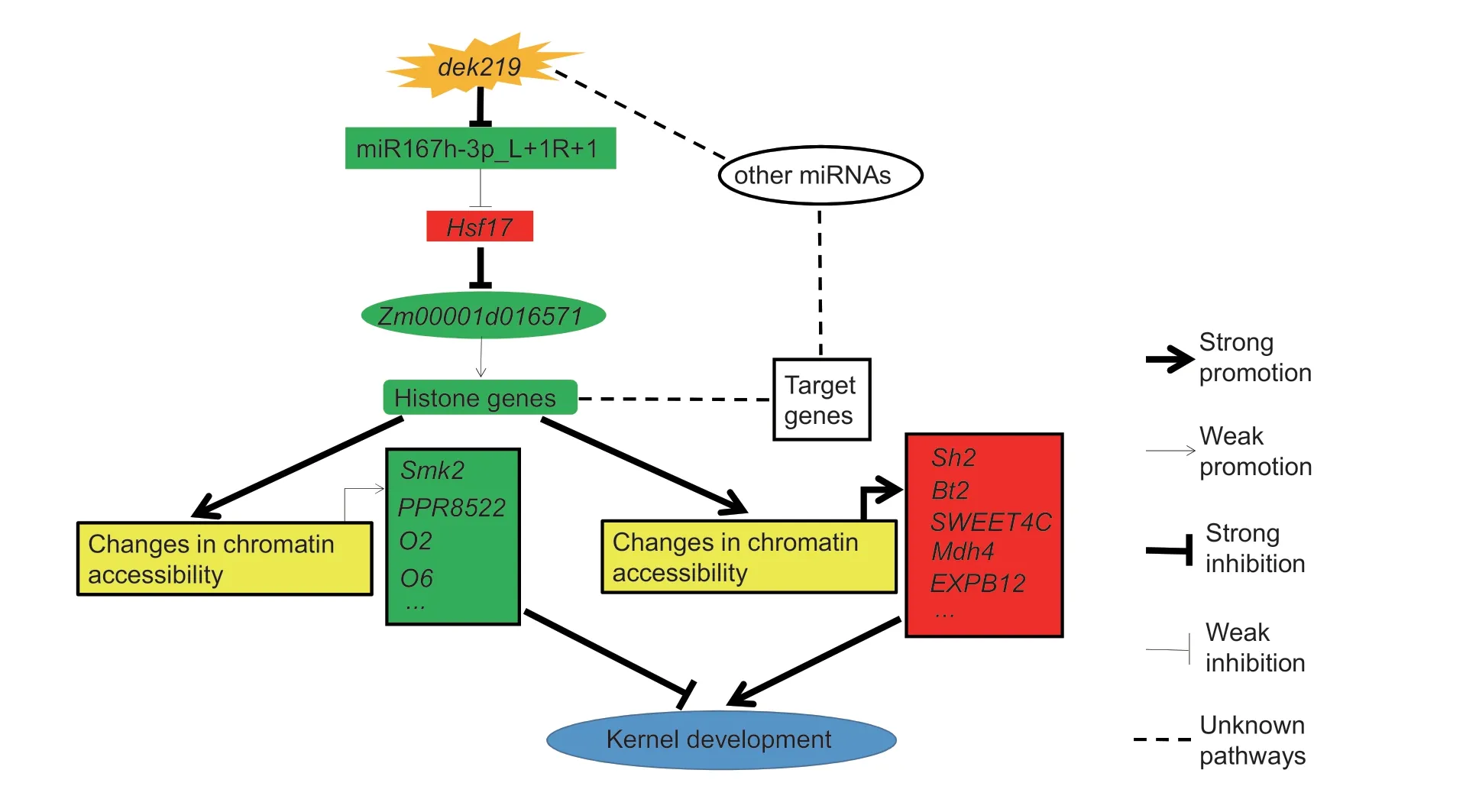
Fig.7 Proposed regulatory model of dek219 in maize kernel development. The red boxes represent up-regulated gene expression,and the green boxes represent down-regulated gene expression.
Acknowledgements
This research was funded by the National Natural Science Foundation of China (32072071) and the National Key Research and Development Program of China(2021YFF1000304).
Declaration of competing interest
The authors declare that they have no conflict of interest.
Appendicesassociated with this paper are available at https://doi.org/10.1016/j.jia.2023.02.024
 Journal of Integrative Agriculture2023年10期
Journal of Integrative Agriculture2023年10期
- Journal of Integrative Agriculture的其它文章
- The association between the risk of diabetes and white rice consumption in China: Existing knowledge and new research directions from the crop perspective
- Linking atmospheric emission and deposition to accumulation of soil cadmium in the Middle-Lower Yangtze Plain,China
- Genome-wide association study for numbers of vertebrae in Dezhou donkey population reveals new candidate genes
- Are vulnerable farmers more easily influenced? Heterogeneous effects of lnternet use on the adoption of integrated pest management
- lnfluences of large-scale farming on carbon emissions from cropping:Evidence from China
- Spatio-temporal variations in trends of vegetation and drought changes in relation to climate variability from 1982 to 2019 based on remote sensing data from East Asia