
质子交换膜燃料电池氧还原Pt基催化剂研究进展
2023-09-14 02:57:08宋学实曲微丽王振波
石油化工高等学校学报 2023年4期
宋学实,曲微丽,赵 磊,王振波
(1.哈尔滨师范大学 化学化工学院,黑龙江 哈尔滨 150025;2.哈尔滨工业大学 化工与化学学院,黑龙江 哈尔滨150001)
几十年来,中国的经济发展为世界所瞩目,工业发展也十分迅速,但全球生态环境的恶化仍在加剧[1]。因此,我国在2021 年提出了“碳中和、碳达峰”等理念。在此时代背景下,新能源的开发和建设尤为重要,其中氢能因为较高的能量密度以及氧化产物的绝对清洁等特性,被认为是未来动力的完美能量来源[2-4]。质子交换膜燃料电池(PEMFC)可以实现由化学能到电能再到机械能的能量转化,相较于内燃机的工作路径减少了热力学转化过程,能源利用率较高。因此,PEMFC 技术的不断提升、工业生产规模的不断壮大,使交通运输行业越来越重视氢能源的开发和利用。在无数科研人员的不断努力下,燃料电池的研究在低温启动性、功率密度、排放指标等领域已经取得了瞩目的成就[5]。但是,催化剂生产成本高昂、电池使用寿命较短等短板,仍是该领域研究的主要阻力[6]。因此,制备出性能高、稳定性好、成本低的燃料电池氧还原反应(ORR)催化剂,才能从根本上合理利用氢能源。本文以此为出发点,归纳了目前燃料电池阴极Pt 基催化剂ORR的研究方向、影响因素以及未来的发展趋势[7-10]。
1 燃料电池阴极反应原理
从动力学反应角度分析,氧气在阴极上的还原速率非常缓慢,这限制了PEMFC 的应用和生产。到目前为止,贵金属Pt 基催化剂由于其较高的电流密度和较低的过电位仍然是首选的阴极材料[7]。阴极的氧还原反应存在4 电子反应和2 电子反应两种途径[11-13]。
(1)PEMFC 阴极在酸性介质中的4 电子氧还原反应途径如下。
(2)PEMFC 电池阴极在酸性介质中的2 电子氧还原反应途径如下。
式中,*代表催化材料的活性位点。
在碱性条件下O2催化中间产物比较复杂,包括O、OH-、O-2、HO-2等,这使ORR 的机理难以确定,推测大致可以分为4 电子反应过程和2 电子反应过程。
(1)4 电子反应过程:O2被完全还原,最终产物为OH-。过程如下:
(2)2 电子反应过程:O2还原不完全,生成中间产物HO-2,而后中间体再分解生成OH-,过程如下:
研究表明,在酸性Pt 基上强氧结合能在一定程度上促进了*O 的形成,即促进了4 电子反应途径。相反,2 电子反应途径的最终产物是H2O2,它会破坏Nafion 膜,造成燃料电池的性能下降[14]。图1 为Pt(111)上的4 电子反应过程和PtHg4上的2 电子反应过程ORR 自由能图[13]。
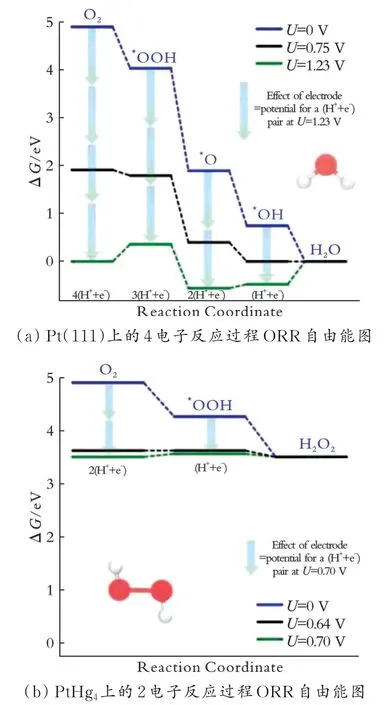
图 1 Pt(111)上的4 电子反应过程和PtHg4上的2 电子反应过程ORR 自由能图Fig.1 ORR free energy diagram of four-electron process on Pt(111) and ORR free energy diagram of two-electron process on PtHg4
由图1 可知,不同金属的ORR 活性图作为*OH结合自由能的函数,对于与含氧物质结合过强的金属,*OH 进一步反应产生H2O 作为基元反应中的决速步骤,而对于与含氧物质结合过弱的金属,O2的活性/解离以及*OOH 的还原程度决定它的反应速率。在这些金属中,Pt 无疑是一个最好的选择。
2 ORR 催化剂催化作用机理
高昂的制造成本只是束缚PEMFC 商业化发展的原因之一,而能否维持催化剂的高催化活性和高稳定性也是重要的影响因素[14]。在阴极的ORR 过程中,2 电子反应途径只需要克服较小的能垒,但中间体过氧化氢有时会逆向反应生成氧气,进而导致中间体不完全转化,使电流效率降低。与此同时,催化剂的活性和稳定性会因为活性中心和碳载体被过氧化氢氧化或腐蚀而降低。相比之下,4 电子反应途径虽然需要克服更高的能垒,但可以实现氧分子转化为H2O 的高效转化,避免中间体过氧化氢的产生。高活性和高稳定性催化剂通常遵循4 电子反应途径。
2.1 动力学影响因素
催化剂表面对含氧中间体(如O*2、*O、*OH、*OOH 和HOOH*)的吸附/解吸过程是ORR 动力学的关键影响因素[15-18]。O2→O*2(O2的吸附过程)作为反应的初始步骤决定后续反应的进程,而反应的初始电位由*OH 的吸附/解吸(高电位下)决定,活性中心的有效数量由*OH 的吸附量决定。更关键的是,催化剂对中间体的吸附能直接影响催化活性,吸附强度过弱会阻碍质子/电子的移动,吸附强度过强会引起脱附。
2.2 催化剂性能促进机制
通过调控催化剂颗粒直径大小和形貌,可以增加催化剂与氧气反应的活性位点的裸露数量,使催化剂的比表面积增大,这种策略是提高氧还原反应性能的有效手段之一[18]。制备核壳结构催化剂以及选用比表面积相对大的催化剂载体可以明显提升催化性能。同时,调整Pt 的电子结构使Pt 的d 带中心下移,从而改变催化剂与有氧物质的结合能,适当调节结合能可从源头上提高催化剂活性。晶面效应、应变效应、配体效应以及Pt 与载体的相互作用是影响Pt 表面电子结构的主要因素[16]。
2.3 催化剂性能抑制机制
Pt 基金属催化剂活性稳定性下降的原因主要有以下两个方面[19]:一方面是Pt 颗粒的溶解、团聚和分离引起有效成分流失;另一方面是催化粒子和载体之间的相互作用因碳载体的腐蚀而减弱。利用制备Pt-过渡金属合金以及制备金属间化合物等方法可稳定催化剂活性。但是,在多次循环测试反应后,Pt 在反应过程中生成PtO,遮盖原有的活性位点;或是其他金属的氧化/溶解导致Pt 层的厚度增加而引起活性下降[20]。相比较之下,碳载体的腐蚀机制比较复杂,载体的石墨化程度、表面积以及材料的多孔结构和表面取向等都是其催化活性的影响因素。因此,在选用不同载体促进催化剂活性的同时,载体的抗腐蚀性也是要重点考虑的因素之一[21-22]。
3 Pt 基催化剂的研究
3.1 合金型Pt 基催化剂
虽然Pt 在ORR 中展现了非常优异的催化性能,但其储量不高、成本高昂的特点很大程度地阻碍了燃料电池的发展。因此,自1980 年首次发现Pt合金可以作为ORR 催化剂以来,这一方法越来越受到 研 究 者 的 关 注[23]。在Pt-M(M 代表Pd、Ni、Co、Fe、Cu、Mn 等)合金催化剂中,过渡金属元素方便获得,储量丰富,成本较低。加入过渡金属M,可实现催化剂的电子和几何结构的良性调控,使d 带中心位置发生偏移,减小与O2反应的结合能,降低贵金属Pt 的加入量,从而实现控制成本的目标[24-27]。
哈尔滨工业大学王振波教授课题组先选用微波辅助二元醇的还原方法制备前驱体,随后在管式炉中煅烧,热还原制备了一种附着在Co-NC 上的合金型Pt-Co催化剂[28]。图2为Co-N-C载体、商用Pt/C催化剂、Pt/Co-N-C 催化剂和Pt-Co/Co-N-C 催化剂的SCV 曲线和H2O2产率图[28]。结果表明,合金化过程使原子Co 进入Pt 原子的晶格中,Co-NC 载体和Pt-Co 合金催化剂之间的相互作用增强,表现出独特的氧还原活性。通过这种方法制备的Pt-Co/Co-NC 催化剂在电化学活性测试中表现出显著的高活性。
结合理论计算结果和相关Pt-Co 合金报道得出,Pt-Co 双位点吸附模式更有利于*OOH 中间体的稳定,*OOH 吸附的稳定性使后续反应中O-O 的断键更加容易;在合金形成过程中,Pt-Co 合金和载体间的层间距随之变化,从而提高了催化剂的稳定性。同时,由于Co 原子的加入,Pt 的电子结构发生了适当的变化,对催化剂的催化性能产生了有效的影响。
T.D.Le 等[29]将Pt-Ni 合金纳米颗粒(NPs)共同沉积在氮掺杂碳(NDC)载体上制备了新型电催化剂PtNiSA-NPs-NDC。图3 为PtNiSA-NPs-NDC 催化剂的ORR 活性与对比样性能对照。由图3 可知,Pt-NiSA-NPs-NDC 显示了最佳的ORR 活性,半波电位为0.912 V,高于商业化的Pt/C 的半波电位(0.857 V)。Pt 与其他过渡金属形成合金的过程可调节中间体的吸附能,进而提升Pt 的ORR 活性,这是因为在合金中存在配体效应和应变效应,引起了Pt 的d带中心下移,通过进一步降低中间体的吸附能来提高其催化性能[30-31]。
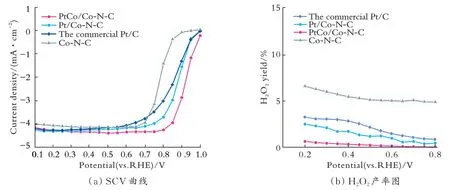
图 2 Co-N-C 载体、商用Pt/C 催化剂、Pt/Co-N-C 催化剂和Pt-Co/Co-N-C 催化剂的SCV 曲线和H2O2产率图Fig.2 SCV curves and H2O2 yield diagram of Co-N-C carrier, commercial Pt/C catalyst, Pt/Co-N-C catalyst and Pt-Co/Co-N-C catalyst

图 3 PtNiSA-NPs-NDC 催化剂的ORR 活性与对比样性能对照Fig.3 The ORR activity of PtNiSA-NPs-NDC catalyst is compared with that of the control sample
在二元合金催化剂显示出特别的ORR 活性的基础上,在二元合金催化剂中掺杂第三种金属所带来的突出优势也被更多研究者关注。X.Q.Huang等[32]在Pt3Ni 二元合金的表面添加金属Mo 得到了三元合金催化剂Mo-Pt3Ni/C。金属Mo 的存在调节了两种金属表面的电子和几何特性,增大了催化剂的稳定性。T.Zheng 等[33]采用电偶置换法制备了Pt-Pd/Ni/C 催化剂,且将利用相同置换方法制备的Pd-Ni/C、Pd-Pb/C 催化剂作为对比样。电化学测试结果表明,三元合金催化剂的ORR 活性和稳定性都高于对比样。分析其主要原因:(1)随着第三种金属的加入,合金中Pt、M 金属的溶解性降低,催化剂性能更平稳;(2)加入第三种元素后,三种金属原子间产生了协同作用,调整了催化剂的电子和几何结构,对增强催化剂活性产生了积极的作用。除了以上提及的几种制备合金纳米催化剂的方法外,还有液相气体还原剂(GRAILS)、湿法热还原等[34-36]。
3.2 核壳型Pt 基催化剂
虽然制备合金型催化剂能够降低催化剂制备成本,但是合金中的金属原子在持续工作下的溶解损耗易降低催化剂的催化活性。当前,制备核壳型Pt 基催化剂是一种比较有效的替代策略[37-40]。核壳型Pt 基催化剂可实现低Pt 负载、低成本以及高耐磨性,具有非常丰富的应用前景。核壳型Pt 基催化剂比较成熟的合成方法有去合金化、金属的热处理和金属的电化学处理等[41]。
去合金化包括电化学去合金化和化学去合金化途径。电化学去合金化通常采用1 mol/L 的H2SO4溶液在80 ℃对前体粉末进行处理,将得到的催化剂溶液冷冻干燥。P.Mani 等[42]提出用脱合金法制备Pt-Cu 核壳结构催化剂:首先,采用浸渍-冷干-热还原的路线制备Pt-Cu 合金;在硝酸铜水溶液中对Pt-Cu 合金浸渍干燥后,在H2氛围下热还原得到前驱体Pt-Cu 合金,以前体溶液为浆料制作燃料电池阴极;将膜电极组件与燃料电池组装后,在脱氧条件下阴极工作200 次,使阴极催化剂实现电化学脱合金化。图4 为通过前体催化剂由伏安法选择性溶解Cu 分步原位制备脱合金Pt-Cu 电催化剂的流程[42]。在催化剂总质量相同的条件下进行电化学测试,对去合金化的Pt-Cu 催化剂与标准Pt 的ORR活性进行了对比,结果表明Pt-Cu 核壳结构的电化学活性远远领先于标准Pt。

图 4 通过前体催化剂由伏安法选择性溶解Cu 分步原位制备脱合金Pt-Cu 电催化剂的流程Fig.4 De-alloyed electrocatalysts were prepared in situ step by step by selectively dissolving Cu from the precursor catalyst of Pt-Cu by voltammetry
G.M.Leteba 等[43]通过共热解方法制备二元Pt-Ni 纳米粒子,优化了合成变量,从而显著提高了催化性能。然而,许多研究发现,Pt 基核壳结构催化剂的形态、局部原子的结构和组成等因素决定催化剂的ORR 活性,所以实现这些因素的可控合成是提高核壳型Pt 基催化剂性能的关键步骤。制备催化剂时通过降低Pt 的用量来实现商业化,造成了催化剂在高电流密度下的性能随之降低,ORR 动能损耗仍然是导致其性能下降的主要原因。F.Zhou等[44]通过一步自组装煅烧工艺合成了具有核壳结构的Pt/C 催化剂,用异丙醇和Nafion 处理商业铂碳;商业铂碳干燥后在N2氛围下煅烧分解形成不含磺酸盐的多孔涂层。结果表明,Pt 颗粒与Nafion 之间存在较薄的多孔层,在不影响导电性和氧气传导速率的情况下,有效地提升了催化剂的性能。
K.Sasaki 等[45]利用低电位沉积(UPD)杂质金属的电流置换(GD)法制备了单层Pt 原子(PtML)的PtML/Pd/C 电催化剂,使用NaBH4还原法制备Cu单层催化剂,通过电位沉积法用K2PtCl4置换单层Cu 得到了该催化剂。这种方法合成的催化剂无论是在性能还是在稳定性上都有良好的表现。
核壳型Pt 基催化剂主要包括两部分,即核层和壳层,核层主要由非贵金属组成,壳层主要由贵金属Pt 层组成。非贵金属核层被贵金属Pt 层包裹,在减少非贵金属损耗降解的同时,实现了降低贵金属的用量、节约制备成本的目的。此外,核层的非贵金属除了单原子,还可以采用双原子核层。核壳结构的优势主要有:(1)Pt 核层与壳层金属之间的相互作用可调节Pt 表面的电子状态;(2)适当的Pt 层厚度可使金属核的抗氧化性和抗溶解性得到明显提升。以上两种优势,一方面提升了ORR 活性,另一方面提高了催化剂的稳定性。总之,调整催化剂形貌、改变金属成分、调节表面取向和增减壳层厚度等,是提升核壳型Pt 基催化剂ORR 性能的有效手段。
3.3 担载型Pt 基催化剂
合理地选择催化剂载体可以有效减少催化剂的团聚,显著地提高催化剂的催化效率。合适的催化剂载体应该具有比表面积大、导电活性好、孔隙率适当、抗中毒能力强、与金属Pt 有一定的协同作用等优点。
3.3.1 金属有机骨架热解产物碳担载Pt 基催化剂 沸石-咪唑-框架-8(ZIF-8)金属有机框架的热解会得到M-NC 催化剂(M 表示金属原子)。在合成过程中,ZIF-8 前体中金属离子的种类、成分和含量、ZIF-8 的颗粒直径、热解温度和热解氛围、升温速率等因素的改变都会影响催化剂的性能[46-47]。
Y.Liu 等[46]研究发现,在羧酸盐(OAc)的作用下,Fe-NC 催化剂可获得丰富的活性中心和新颖的传质途径,他们将羧酸盐的作用称为“分子剪刀”;经过裁剪后的Fe-NC-OAc 催化剂在电化学ORR 测试中半波电位达到了0.838 V,动力学电流密度为15.87 mA/cm2,表现出较高的催化性能,已经接近于近期关于Fe 基催化剂研究的最高水平,甚至接近商业铂碳的催化性能。除Fe-NC 催化剂外,课题组成员还制备了Co-NC[28]、Ru-NC[48]等催化剂载体,这些载体不仅可有效地降低Pt 金属的用量,而且还能稳定地提升催化剂的性能。
3.3.2 石墨烯及类石墨烯担载Pt基催化剂 石墨烯是碳原子以sp2杂化方式形成的六角形单层片状单质。石墨烯纳米片具有超大的π-电子共轭体系、超高的理论比表面积和优异的电子导电性,自2004年从石墨中分离出后,石墨烯材料作为催化剂载体在多篇报道中被提及[49-52]。提升催化剂性能的有效方法之一是使氧还原反应产物快速到达催化剂表面,从而能够高效地利用各个催化位点。因此,除催化剂自身性能、形貌等影响因素外,对催化剂载体的结构以及形态进行改善设计也是较有发展前景的研究方向。
L.Sun 等[53]以冻干氧化石墨烯、甲醇、二甲基咪唑和三乙胺为原料,通过原位控制在不同尺寸的氧化石墨烯表面还原Co-ZIF 前驱体,采用热还原的方法得到了氮掺杂石墨烯担载Pt-Co 合金催化剂。结果表明,该催化剂的半波电位高达0.86 V。
Z.Zhang 等[54]在类石墨烯结构的MXene 材料中添加凯琴黑(Ketjen Black),凯琴黑被适当地填充在MXene 夹层之间,从而合成了具有三维立体结构的材料(MCM),随后采用湿化学方法用甲酸将氯铂酸还原制得Pt/MCM 三维催化剂。性能测试及物理表征结果表明,MXene 层与层之间的狭小缝隙确实存在,因为凯琴黑的夹杂,催化剂的导电性和传质均得到补强,进而催化剂的活性得到提高。与此同时,这种三维结构使催化剂的稳定性得到提高,这是因为该结构可有效调整层状MXene 材料在作为催化剂载体时由自身出现的堆叠造成降低催化剂活性这一缺陷。
本节提到的两种催化剂载体均为碳质载体,常见的碳质载体包括炭黑、碳纳米管、碳纳米纤维等。S.Wang 等[55]制备了炭黑和还原氧化石墨烯协同作用的复合催化剂载体。除碳质载体外,较常用的催化剂载体还包括非碳质载体和导电聚合物等[56-57]。近几年,非碳质载体中形成的金属间化合物,如Pt-Co 金属间化合物以及Pt-Ni 金属间化合物等,因其可提高催化剂的耐中毒性而受到研究者的广泛关注[58-59]。
3.4 单原子Pt 基催化剂
节约催化剂的制备成本,除降低Pt 的负载量外,保证Pt 金属的高利用率也是高效电化学催化剂的必备品质[60-61]。通常情况下,Pt 金属颗粒半径越小,Pt 的利用率越高,Pt 颗粒变成簇或是以单原子形式存在是最理想的参与方式,在ORR 过程中会发现小尺寸Pt 原子的参与。
J.Liu 等[60]报道了一种将碳缺陷锚定Pt 单原子负载在炭黑上的电化学催化剂,在200 ℃下将炭黑放置在含一定量H2O2的密封乙醇溶液中进行热处理,H2O2的存在使碳的表面产生缺陷,而正是这些缺陷优化了碳载体上催化剂的分布,进而使催化剂的催化活性和稳定性均显著提升。电化学测试结果表明,与Pt/炭黑催化剂相比,该Pt/BP 催化剂ORR 性能表现优异,并且Pt 的利用率达到0.09 g/kW,根据物理表征以及密度泛函理论计算推测,催化剂的活性中心以碳原子锚定的单原子Pt 为主。
J.Liu 等[61]报道了一种新颖的N 锚定Pt 单原子电催化剂。该催化剂采用醇的热还原和高温热解还原相结合的优化制备方案,以三聚氰胺、炭黑、Pt(acac)2等为原料,在制备过程中经历两次煅烧后重建Pt 原子的配位环境,实现了被N 锚定的Pt 单原子催化剂。这是因为N 杂原子的掺杂促进了Pt 原子的分散,同时与Pt 相邻N 原子的配位环境在高温条件下被重建,吡啶基N 锚定单原子Pt 催化剂遵循4 电子反应途径,同时在锚定过程中生成活性位点。总之,近年来随着对Pt 基催化剂的逐渐开发,因单原子Pt 的较高利用率,单原子Pt 基催化剂被认为是未来具有较高开发潜力的催化剂之一。
4 总结与展望
介绍了燃料电池阴极反应原理及ORR 催化剂催化作用机理,总结了PEMFC 阴极氧还原反应Pt基催化剂的研究方向,并从合金型Pt 基催化剂、核壳结构Pt 基催化剂、不同载体担载Pt 基催化剂、单原子Pt 基催化剂4 个方面详细阐述了目前催化剂研究的主要方向。随着我国整体科研能力的高速发展及物理表征手段的日益丰富,虽然可以借助原位表征技术等手段精确地制备和构筑一些理想的催化剂,但是对其在理论计算领域的研究仍然有一定的局限性。理论计算在一定程度上可帮助研究人员从本质上探究催化剂的工作机理,因此,未来对Pt 基催化剂理论计算和实验相结合的研究能力仍需提高;同时,PEMFC 在商品化发展的现阶段仍处于瓶颈期,优化工艺、降低成本等仍是制备高活性、高稳定性的Pt 基催化剂中迫切需要解决的问题。
猜你喜欢
少儿科学周刊·儿童版(2021年22期)2021-12-11 21:27:59
少儿科学周刊·儿童版(2021年22期)2021-12-11 21:27:59
少儿科学周刊·儿童版(2021年22期)2021-12-11 06:42:32
粉末冶金技术(2021年3期)2021-07-28 06:26:32
原子与分子物理学报(2021年2期)2021-03-29 07:30:58
中国有色金属学报(2018年2期)2018-03-26 07:58:39
西安工程大学学报(2016年6期)2017-01-15 14:08:22
焊接(2016年8期)2016-02-27 13:05:16
湖北师范大学学报(自然科学版)(2015年1期)2016-01-10 08:41:29
无机化学学报(2014年10期)2014-02-28 17:33:24
