
CO2RR 稀土基催化剂的研究进展
2023-09-14 02:56:54刘嘉敏越婷婷郭少红贾晶春贾美林
石油化工高等学校学报 2023年4期
刘嘉敏,越婷婷,常 迎,郭少红,贾晶春,贾美林
(内蒙古师范大学 化学与环境科学学院,内蒙古 呼和浩特 010022)
随着现代工业的迅速发展,人类大量燃烧煤和石油等化工燃料。但是,化工燃料在燃烧时会释放大量的CO2,而CO2的过度排放会导致全球变暖、海平面上升、温室效应加剧等一系列环境问题。CO2的过度排放带来的环境问题,已引起研究人员的广泛关注,一方面可通过改变能源供给结构、减少化工燃料的使用来降低CO2排放量[1],另一方面可通过将CO2转化为高附加值燃料或化学品以减少CO2排放量,从而缓解环境问题。目前,降低CO2排放量的方法有通过电催化、热催化、光催化等方法将CO2转化为高附加值燃料或化学品。但是,热催化CO2还原反应对操作压力和反应温度有较高的要求[2-4],光催化CO2还原反应具有效率较低[5-7]、重现性差[8]等缺点,而电催化CO2RR 具有能够在常温常压下进行反应、通过调节外加电位可精确控制反应过程、电解液能够重复利用以及转化效率高等优点,因此得到了广泛关注,被认为是一项有潜力的技术。
在过去的研究中,针对电催化CO2RR 开发了一系列电催化剂,如贵金属[9-11]、过渡金属[12-14]、金属有机骨架材料[15-17]、碳基材料[18]和氮掺杂的碳基材料[19-21]等催化剂。但是,目前的电催化剂仍然存在成本高昂、合成方法复杂、催化效率低、选择性差等特点,因此设计和开发高选择性、高催化效率的CO2RR 电催化剂迫在眉睫。研究人员对CO2RR 稀土基催化剂进行了深入的研究。例如,P.Chen 等[22]利用La 独特的4f 和5d 轨道与N 原子之间进行轨道杂化,形成电荷转移通道,从而促进了光催化CO2还原反应中CO2的活化、*COOH(*为反应中间体的标志,下同)的形成和*CO 的解吸。因此,稀土元素催化剂在电催化CO2RR 中也具有一定的潜在作用,需要从材料结构、催化剂设计等方面认识稀土元素催化剂,从而使其得到更好的利用。
1 CO2RR 原理和基本参数
1.1 CO2RR 原理
CO2分子具有稳定的线性结构,其中C=O 的键能高达750 kJ/mol[23],因此需要施加外加电位才能活化CO2分子;其次,在电催化CO2RR 过程中还伴随着多个质子和电子的转移[24],会导致反应速率缓慢,所以需要高效的电催化剂来催化CO2RR。当采用的电催化剂不同时,所得产物也不同,总结了标准实验条件(pH 为7、温度为25 ℃、压力为1.01×105Pa、电解质浓度为1 mol/L 的电解液)下电催化CO2RR 中 不 同 产 物(CO、HCOOH、CH4、C2H4、HCH2OH 和CH3CH2OH)[25-26]在水溶液中的电极电位(相对于标准氢电极RHE),结果见表1。电催化CO2RR 过程主要分为3 步:催化剂将CO2吸附在其表面并活化CO2;通过质子/电子转移形成反应中间体;中间体从催化剂表面脱附形成产物或参与下一步反应。

表1 标准实验条件下CO2RR 半反应的电极电位[25-26]Table 1 Electrode potential of CO2RR half reaction under standard experimental conditions[25-26]
在电催化CO2RR 过程中,催化剂通过控制反应中间体在催化剂表面的吸附能控制产物的选择性。一般认为,Au、Ag、Zn、Ga 等催化剂的产物为CO,Pb、In、Sn、Bi、Cd、Bi 等催化剂的产物为HCOOH,Pd、Co 等 催 化 剂 的 产 物为CH3OH[25];Cu 基 催 化 剂是唯一一种能够将CO2还原为CH4、C2H4等碳氢化合物和CH3OH、C2H5OH 等碳氧化合物的高选择性的催化剂[27-28]。电催化CO2RR 的产物可分为C1产物(CO、HCOOH、CH4、CH3OH 等)和C2产 物(C2H4、C2H5OH 等)。生成C1产物的过程见图1 的上半部分。电催化CO2RR 的步骤:将一个电子(e-)转移到CO2反应物上形成CO2阴离子自由基中间体(*CO-2);*CO-2通 过质子(H+)转移形成*COOH、*OCHO、*HOCO,三种中间体与H+和e-结合形成HCOOH;*HOCO 与H+结合释放H2O 并形成*CO,在催化剂表面脱附释放CO[29]。如果催化剂对*CO的吸附力很强,则析氢反应(HER)占主导地位;如果催化剂对*CO 的吸附力较弱,则*CO 会从催化剂表面脱附而生成CO;只有当催化剂表面对*CO 具有适宜的吸附力,才能进行进一步的还原反应形成CH4、CH3OH[30]。生成C2产物的过程见图1 的下半部分。通过*CO 的二聚促进C—C 偶联的发生,并生成*OCCO,通过质子电子转移[31-32]形 成C2H4和C2H5OH。
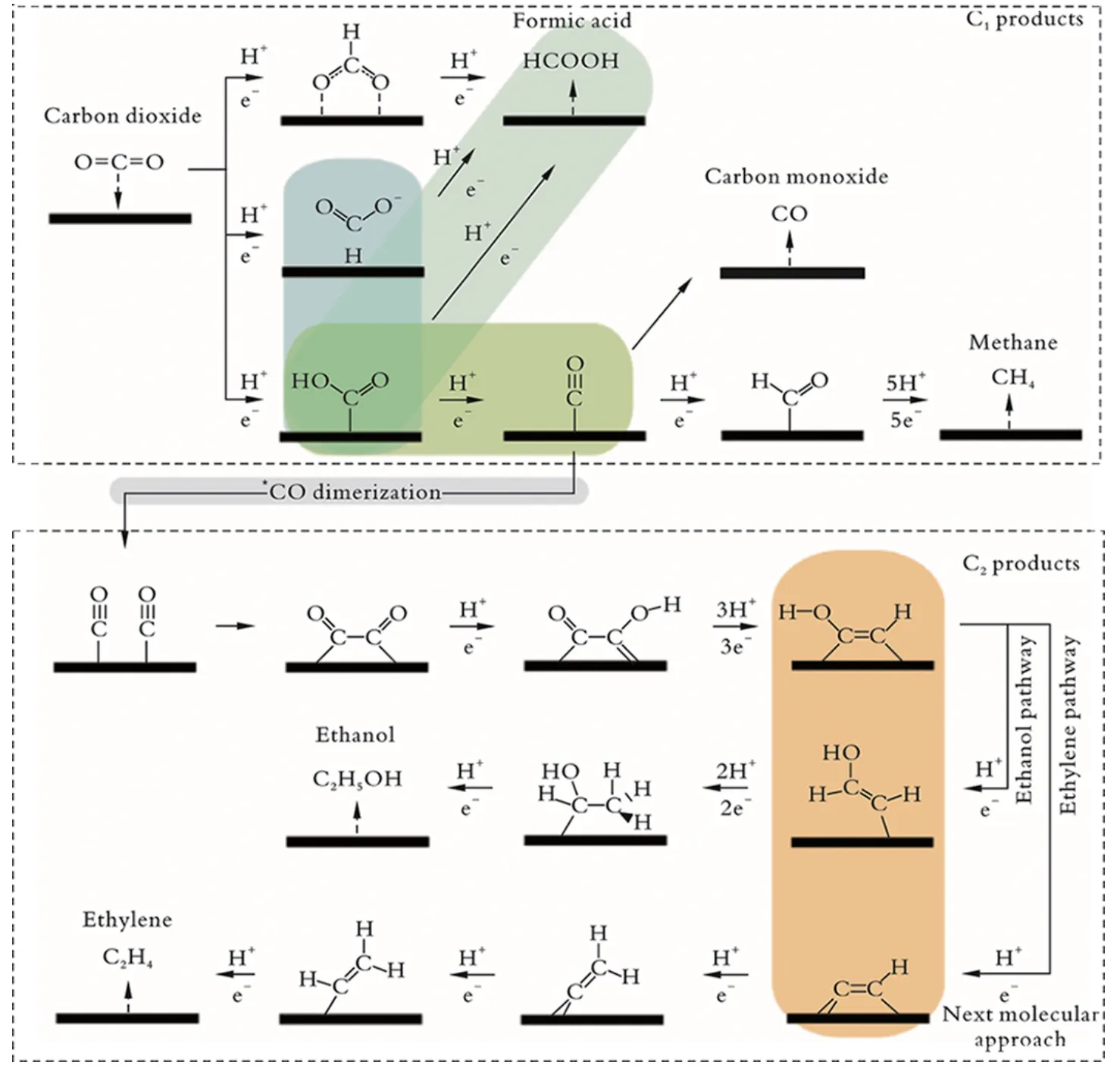
图1 C1/C2产物的生成过程[33]Fig.1 Product generation process of C1/C2[33]
电催化CO2RR 的反应涉及多个质子和电子的转移,而复杂的反应过程会使其产物选择性降低。但是,在电催化CO2RR 中,生成CO 的反应只涉及两个e-和两个H+的转移,不仅受到的动力学影响较小,而且反应过程较为简单。电催化CO2RR 反应发生在水性电解质中,因此同时伴随着HER 的发生。由于HER 的电位范围与CO2RR 相似,因此HER 常与CO2RR 形成竞争反应。通过控制CO2RR 和HER,可调控CO 和H2的比例,提高CO 和H2的混合气(合成气)的选择性,将CO2直接转化为有价值的化学品或燃料[33-35]。传统的合成气制备方法有CH4-CO2重整、水煤气变换(RGWS)等,其反应过程较为复杂,实验条件苛刻[36]。电催化CO2RR 制备合成气反应过程简单,成本较低,并且能够大规模应用,但存在反应过电位高、无法在高电流密度及宽电位范围调控合成气中CO 与H2比例的缺点[37]。过渡金属催化剂N-FE3C/rGO 通过控制n(CO)/n(H2)为1∶2,用于费托法合成甲醇;过渡金属催化剂Co@CoNC还原CO2生成n(CO)/n(H2)为1∶2 的合成气,用于合成醇类或烯烃[38-39]。稀土元素既可作为HER 的活性位点,也可作为CO2RR 的活性位点,同时具有降低过电位、增大电流密度的作用[40],为制备合成气提供了一种新型的催化材料。稀土元素具有与过渡金属相似的电子结构,并且具有多电子轨道,能够通过镧系收缩改变周围原子的电子结构,实现合成气的可控生成。利用稀土元素制备合成气,不仅能够提高稀土元素资源的利用率,而且能够利用其独特的结构实现“碳中和”及“碳循环”。
1.2 CO2RR 的基本参数
CO2还原性能参数包括法拉第效率(FE)、电流密度(j)、稳定性、过电位和转化频率(TOF)。
法拉第效率(FE)[41]:FE 表示电催化剂的选择性,与催化剂的活性位点、反应途径和反应中间体的吸附能有关。FE 的表达式为:FE=ZnF/Q。其中,Z为生成产物转移的电子数;n为生成产物的物质的量,mol;F为法拉第常数,取值96 485 C/mol;Q为输入电荷,C。
Z.Y.Zhao 等[42]利 用CeO2修 饰Cu 电 极,在电位为-1.05 V 时生成C2产物的FE 为61%,而未修饰的Cu 电极生成C2产物的FE 仅为23%。这是因为:CeO2诱导Cu 表面产生了CuOx,并利用CeO2与Cu 的界面进行非静态混合,促进了C—C 耦合,从而提高了C2产物的选择性。
电流密度(j)[43]:j是在一定电位下通过电极几何面积归一化测得的总电流,表示CO2转化为还原产物的速率,主要与电子和质子转移速率和系统阻抗有关。当FE 相近时,j高的催化剂其产物产率较高。
过电位[44]:过电位通过CO2RR 半反应的标准电位与外加电位的差值进行计算。CO2RR 是一个非自发的过程,在实际电催化条件下,CO2RR 半反应需要比标准电位更负的电位驱动。
稳定性[45]:恒定的FE 和j表示催化剂能够在长时间、恒电位下保持选择性和活性的能力。很多催化剂在经过长时间使用后,由于催化剂的活性位点失活和不可逆的结构变化,导致其活性衰减。稀土元素具有独特的4f 轨道和大尺寸,因此其与载体的相互作用强,结构不容易发生变化,能够增强催化剂的稳定性[46]。
转化频率(TOF)[47]:TOF 是表征催化剂内在活性的一个参数,其表达式为:TOF=特定产物的物质的量/(活性位点的物质的量×时间)。目前,单原子催化剂的转化率接近100%。C.Z.He 等[48]利用稀土元素Sc 单原子修饰了单分子膜P2Si,结果表明修饰后的分子膜可形成稳定的单原子催化剂,有利于提高CO2RR 的转化率。
2 CO2RR 稀土基催化剂
稀土元素由15 种镧系元素(La-Lu)以及与镧系元素有相似化学性质的钇(Y)和钪(Sc)组成,其电子排布式为[Xe]4fn-15d16s2或[Xe]4fn6s2(除Y、Sc外)。Y、Sc 的电子结构排布式分别为[Ar]3d14s2和[Kr]4d15s2。独特的电子结构使稀土元素具有独特的性质。稀土元素6s 轨道上的两个电子和5d 或4f轨道上的一个电子容易丢失[39],因此大多数稀土离子的化合价为+3。但是,在某些化合物中也可以发现变价的稀土离子,如Ce4+和Pr4+。这是因为:经过变价的稀土离子,在洪特规则的前提下,当其d轨道、f 轨道处于全空位(d0、f0)或全充满(d10、f14)时,结构更稳定[49]。稀土元素因具有独特的电子结构和价态可变的特性,在作为主催化剂、载体和助催化剂时可发挥独特的作用。
2.1 稀土基主催化剂
在整个CO2RR 过程中,需要电催化剂催化反应的进行,催化剂的活性位点决定产物的选择性[50]。传统催化剂通常只有部分表面的原子作为活性位点,而大多数亚表面的原子不能直接参与反应[51]。单原子催化剂的原子利用率接近100%[52],已引起研究人员的关注。目前,大多数研究主要集中在过渡金属单原子催化剂上,该类催化剂通常由过渡金属(M)与配位阴离子(N、P、S 等)构成,呈M-N4的电子结构[53]。该结构使大多数金属位点暴露在催化剂表面,能够产生丰富的活性位点,从而有利于电子转移[54]。目前,对稀土元素单原子催化剂进行的研究较少,因此本文主要介绍用于CO2RR 的稀土元素单原子催化剂。稀土元素与过渡金属的不同点:稀土元素的原子尺寸较大,独特的4f 轨道使稀土元素更容易与载体中的阴离子发生杂化,使电子离域,从而更容易形成稳定的高配位结构[55]。一般而言,配位数越高,中间体在单原子催化剂上的吸附越弱,更容易将CO2还原为CO。因此,探索稀土元素在CO2RR 中的催化性能具有重要意义。
由于Y 和Sc 的多壳层电子参与成键,反应物分子对稀土元素的强吸附导致其难以在室温下释放反应物分子,因此Y 和Sc 氧化物的纳米材料在室温CO2RR 中没有活性。J.Y.Liu 等[56]以ZIF-8 为模板,将Y、Sc 掺入模板中,经过煅烧得到了氮掺杂碳(NC)的单原子催化剂(Y/NC 和Sc/NC)。通过N和C 与Y、Sc 的配位,使Y 和Sc 的电子结构发生改变,形成了稳定的Y-C3-N3、Sc-N6结构(见图2(a))。图2(a)中,灰色、粉红色和红色球体分别代表C、N 和Y)。由小波变换图(WT-EXAFS)分析结果可知,在Y/NC 中Y 与C、N 进行配位(见图2(b))。由X 射线近吸收边光谱(XANES)分析结果可知,Y/NC 中Y的平均价态为+3,与Y2O3一致(见图2(c))。与N、C配位的Y/NC 和Sc/NC 具有良好的CO2还原性能,其FE(CO)最高为88.3%(见图2(d))。
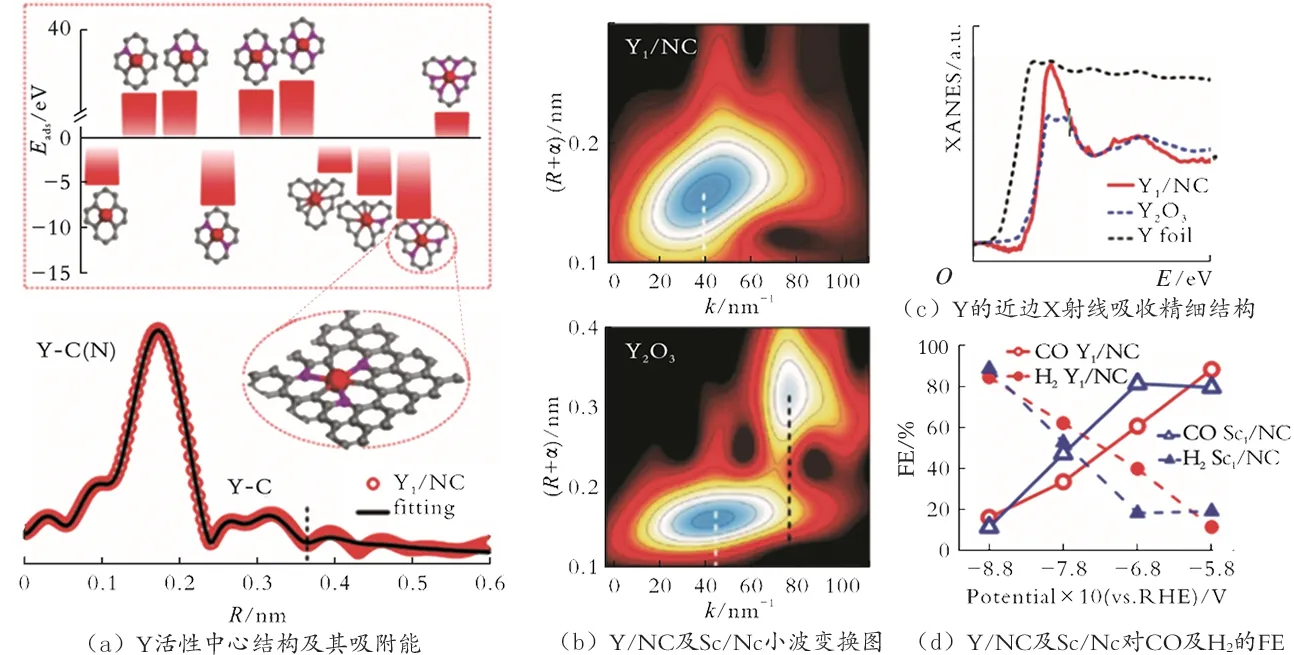
图2 Y/NC 及Sc/NC 的电子结构和电化学性能测试图[56]Fig.2 Electronic structure and electrochemical performance test diagram of Y/NC and Sc/NC[56]
F.Z.Hu 等[57]利用级联锚定策略,在700、800、900 ℃的温度下,将Pr 单原子负载在石墨碳上,制备了催化剂,分别记为Pr-NC-7、Pr-NC-8 和Pr-NC-9,金属负载量(质量分数,下同)为5.07%。Pr 的电子排布式为[Xe]4f36s2,Pr 具有不同的价态,因此Pr 基催化剂具有良好的CO2储存和活化能力。Pr 的富电子轨道使其电子结构更容易发生改变。由于Pr原子尺寸较大,Pr 倾向与NC 形成6 配位。通过密度泛函理论(DFT),计算了NC 配位数不同时Pr 的配位能(Ec),结果可知所有结构的Ec 均为负值,但形成PrN6、PrC3N3和PrC3N3-1 结构的Ec 最低,表明这三种结构都能稳定存在(见图3(a))。通过比较PrN6、PrC3N3和PrC3N3-1 的UL(CO2)-UL(H2)的 值(热力学极限势差)和生成*COOH 的自由能发现,PrN6的UL(CO2)-UL(H2)和生成*COOH 的自由能均最小,表明PrN6结构具有较高的CO 选择性(见图3(b)及图3(c))。根据态密度(DOS)分析可知,Pr 原子的引入会增加费米能级,降低能带间隙,从而提高CO2的电导率和活性(见图3(d))。因此,当Pr-NC-8结构为PrN6时,结构最稳定。Pr-NC-8 中含氮量较高,可形成较多的M-N 活性位点,导致其在电位为-0.43 V 时FE(CO)较高,其值为93%。因此,可利用阴离子与稀土元素的富电子轨道配位,调节稀土元素的电子结构,使其具备催化CO2RR 性能,也可以通过调节配位数控制CO2RR 过程中中间体的形成,从而进一步控制产物的生成。
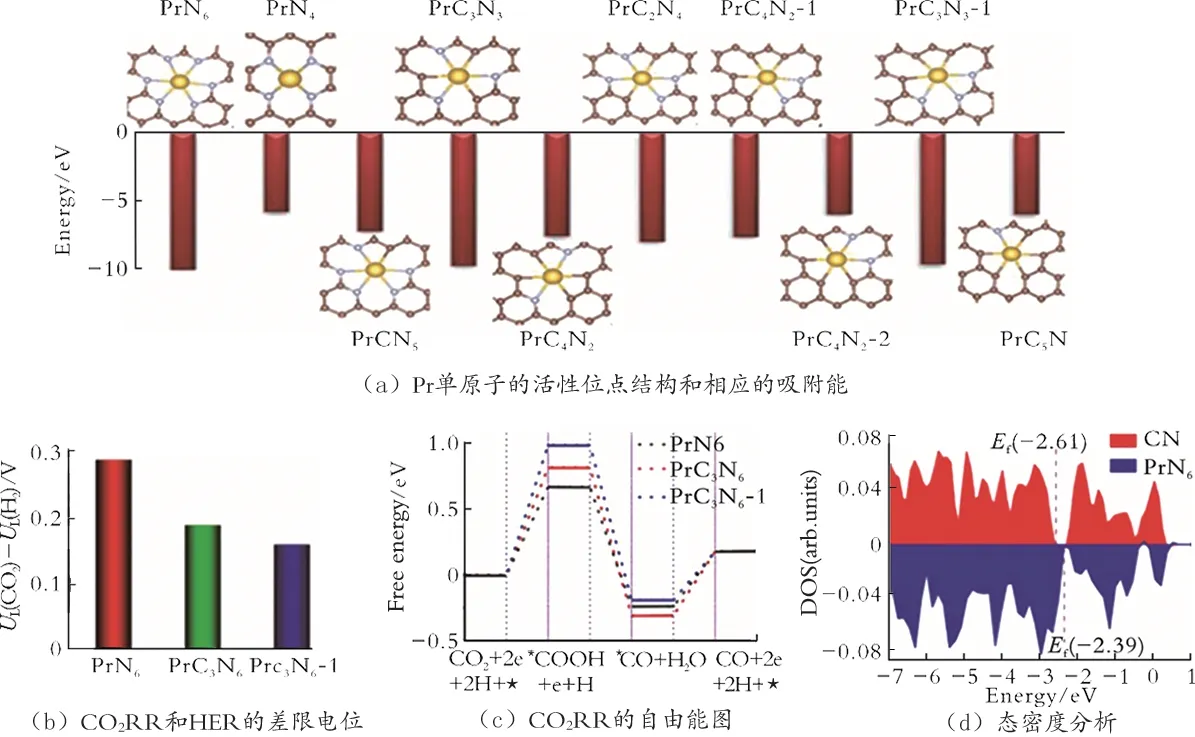
图3 Pr-NC-8 的电子结构和DFT 计算结果[57]Fig.3 Electronic structure and DFT calculation of Pr-NC-8[57]
2.2 稀土氧化物作为载体协同催化
目前,大多数非均相催化剂均由载体和活性组分组成[58]。其中,载体不仅可降低成本,还可通过与其他金属的相互作用,控制活性组分的粒径和形态结构,从而提高催化剂的活性和稳定性[59]。这些特性使稀土氧化物特别是CeO2成为多相催化常用的载体材料。CeO2是一种典型的稀土氧化物,Ce 元素的电子结构排布式为[Xe]4f15d16s2,具有未充满电子的4f 轨道。在还原的条件下,CeO2能够将Ce4+转化为Ce3+,因此CeO2还能起电子“储存器”的作用[60],每一个氧空位的生成,表明有两个Ce4+变为Ce3+。
2.2.1 氧空位 CeO2中Ce4+与Ce3+之间的可逆转换,伴随着氧空位的生成与消除。氧空位的生成不仅可以增加催化剂的活性位点,还可以促进电荷转移,加强CO2的还原性能。当CeO2表面失去一个氧原子时,会形成氧空位,该空位包含额外的电子[61]。这些电子可以被邻近的金属位点获得,而不是在表面上离域,从而有利于CO2的吸附和活化[62]。对CO2RR 而言,通过提高催化剂对CO2反应物的吸附,可提高CO2RR 的转化率。CeO2氧空位通常通过掺杂、还原或缺氧环境下的热处理、引入金属/金属氧化物等处理来产生[63]。
F.Jiang 等[64]利用水热法制备不同形状的CeO2并将其作为Pd 金属的载体,通过实验发现棒状2Pd/CeO2表面的氧空位浓度和数量最高。在H2气氛下,Pd 通过提供游离的H 原子促进CeO2表面氧的消除,从而促进氧空位的形成。楚森林等[65]利用水热法合成了具有(100)、(110)和(111)晶面的CeO2,并利用超声波促进沉积法,将Cu 负载在CeO2上,制备了具有不同晶面的Cu/CeO2。在Cu/CeO2中,Cu+含量在Cu/CeO2(110)、Cu/CeO2(100)和Cu/CeO2(111)中是逐渐减少的。通过电化学测试可知,在电位为-1.05 V 的条件下,Cu/CeO2(110)生成C2H4的FE 和电流密度分别为39.1% 和2.5 mA/cm2,而Cu/CeO2(100)生成C2H4的FE 和电流密度分别为31.8%和2.2 mA/cm2,Cu/CeO2(111)生成C2H4的FE 和电流密度分别为29.6% 和1.7 mA/cm2;Cu/CeO2(110)不仅可促进CO2的吸附,而且使Cu+稳定,从而促进C2产物的生成。
Y.X.Duan 等[66]利用原位电还原法合成Bi/CeOx催化剂,并通过Bi/CeOx催化剂在水溶液中还原CO2制备了HCOOH。无定形的CeOx具有丰富的不饱和结构,能够形成较多的氧空位,因此产生较高的n(Ce3+)/n(Ce4+)。随着CeOx中Ce3+含量的增加,可以形成更多小粒径的Bi 活性位点,从而加强对*HCOO 的吸附,进一步提高CO2RR 的催化性能。S.L.Ning 等[67]通过水热法合成Sn 掺杂的CeO2纳米棒(Sn-CeO2),并考察了Sn 质量分数对催化性能的影响。结果表明,Sn 质量分数为10%的催化剂(Sn-CeO2-10%)性能最佳,其生成HCOOH 的最高FE为81.1%。这是因为Sn-CeO2通过Sn 的掺杂在CeO2(111)表面形成氧空位,降低了反应能垒,促进了HCOOH 的形成。X.L.Zhou 等[68]利用静电纺丝技术,将Cu2+引入CeO2晶格中,得到了Cu-Ce-Ox固溶体。由于CeO2中的Ce4+比Cu2+更容易被还原,因此可避免电子在Cu2+活性位点上聚集,使Cu2+稳定存在。Cu2+的稳定存在能够提高Cu 基催化剂的选择性(见图4)。Cu-Ce-Ox固溶体生成CH4的FE 为67.8%,生成C2H4的FE 为3.6%。因此,利用CeO2作为载体产生氧空位,不仅能加强催化剂对中间体的吸附能力,而且能降低中间体的反应能垒,同时还能保护其他金属,提高CO2RR 的选择性。
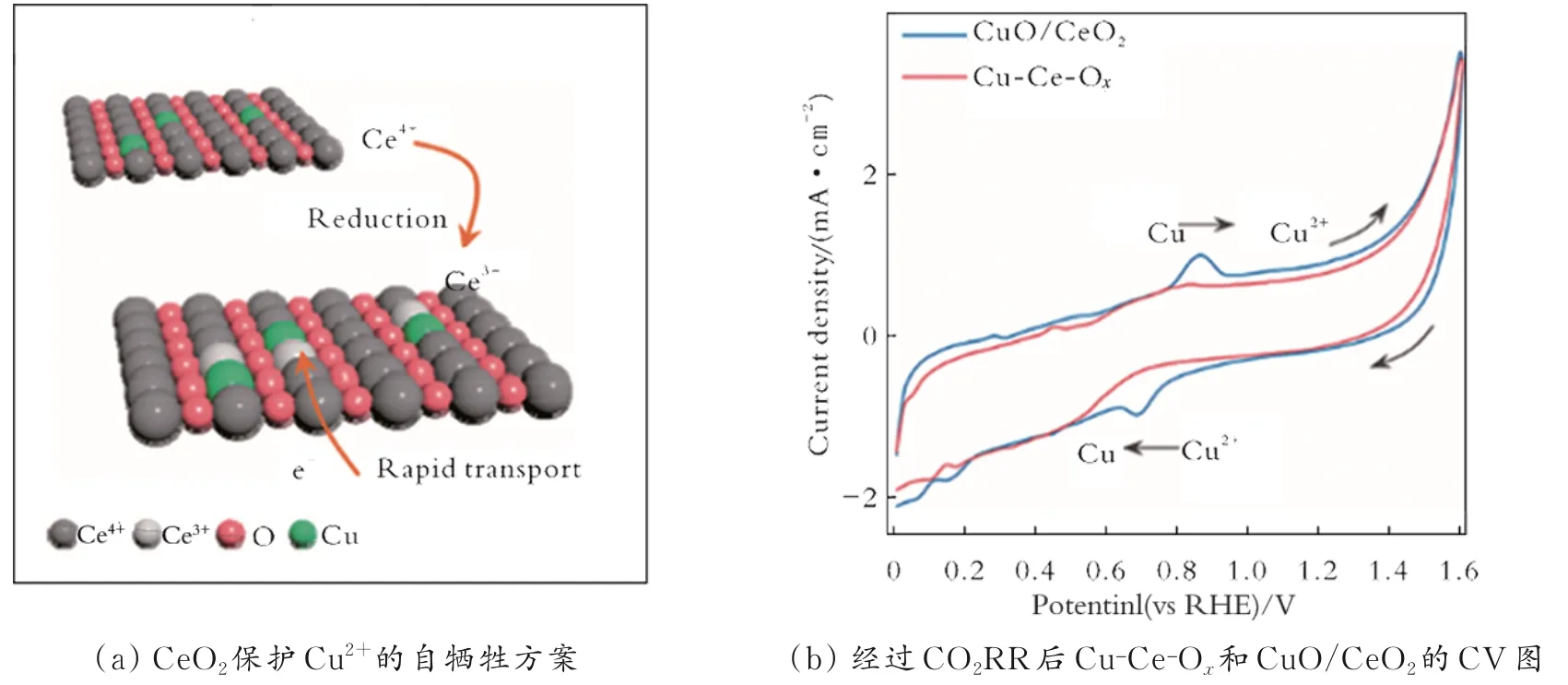
图4 CeO2保护Cu2+的自牺牲方案和在CO2RR 过程中的变化[68]Fig.4 CeO2 protects Cu2+ from Self sacrifice schemex and its changes in the process of CO2RR[68]
2.2.2 CeO2的界面效应 在多相催化CO2RR中,通过金属与载体之间的相互作用形成界面,此界面不仅可提供大量的活性位点,而且还可提高选择性[69]。CeO2作为典型的稀土氧化物,可与其他金属形成复合催化剂,进而催化CO2RR。
L.P.Song 等[69]利用静电纺丝技术合成了CeO2/Bi3NbO7。CeO2的引入降低了CeO2/Bi3NbO7的结晶度,CeO2和Bi3NbO7的界面上存在紧密的原子耦合,这种界面效应使催化剂的价带和导带之间能垒变小,从而改变了CeO2和Bi3NbO7的电子结构,并且促进了CeO2/Bi3NbO7表面上的电子转移,从而有利于生成HCOOH。S.B.Varandili 等[70]利用胶体种子生长法合成Cu/CeO2-xHDs 催化剂,通过Cu 与CeO2之间的界面相互作用,促进了CeO2的还原。结果表明,在电位为-1.2 V 的条件下,Cu/CeO2-xHDs生成CH4的FE 为54.0%。X.Zong 等[71]合成了Cu/CeOx@CNFs 催化剂。结果表明,Cu-CeOx界面不仅可以促进*COOH 的吸附,还可以阻断部分Cu 位点,降低C—C 耦合的机会,从而提高C1产物的选择性;在电位为-0.6 V 的条件下,催化剂 Cu/CeOx@CNFs-2(n(Ce)/n(Cu)=2)生 成CO 的FE 为85.2%。D.F.Gao 等[72]合成了Au-CeOx/C 催化剂,其中Au-CeOx界面是CO2活化和还原的活性位点。结合高分辨扫描隧道显微镜分析可知,当CeOx/Au(111)表面暴露于CO2中时,CeOx的边缘对CO2进行吸附,吸附CO2的位置随着CeOx的边缘分解逐渐向CeOx的中心转移,从而促进CO2的吸附与活化(见图5(a))。根据DFT 计算结果可知,Au-CeOx降低了生成*COOH 的自由能,从而促进了CO2RR(见图5(b))。在电位为-0.89 V 的条件下,Au-CeOx/C 生成CO 的FE 达 到89.1%,远 高 于Au(59.0%)和CeOx(9.8%)。C.W.Lee 等[73]通过烧结纳米晶体制备了Cu/CeO2催化剂。结果表明,CeO2可削弱相邻Cu 位点的氢键能,并通过与*OCCO 的氧原子之间的化学作用稳定*OCCO 中间体,从而促进了C—C 耦合反应的发生(见图5(c)、(d))。综上可知,Cu/CeO2和Cu/CeOx@CNFs 都通过界面作用,提高C1或C2产物的选择性。
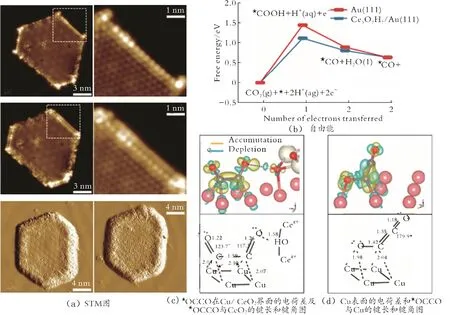
图5 CeOx/Au(111)与CO2之间的相互作用和Cu/ CeO2还原CO2的DFT 计算[72-73]Fig.5 Interaction between CeOx/Au (111) and CO2 and DFT calculation of Cu/CeO2 reducing CO2[72-73]
Z.S.Sun 等[74]利用一锅法合成了Ag/Ag 掺杂CeO2(Ag/CeO2)纳米复合材料。由于Ag+的离子尺寸比Ce4+大,因此添加Ag+会引起晶格膨胀,在CeO2晶格中引入较大的界面面积和大量的氧空位(见图6(a))。结果表明,在电位为-1.01 V 的条件下,Ag/CeO2生 成CO 的FE 为99.0%。 经 过CO2RR 后,Ag 与CeO2界面的相互作用使表面Ce3+保持稳定。
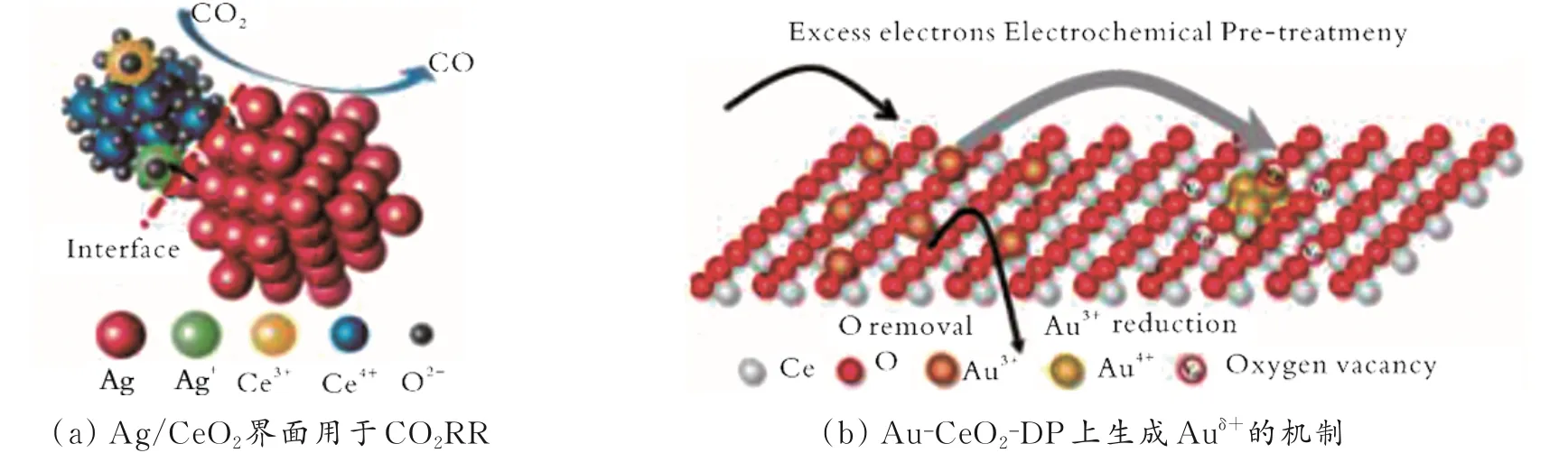
图6 Ag/CeO2 和Au-CeO2-DP 加速CO2RR 的机理[74-75]Fig.6 Mechanism of Ag/CeO2 interface and Au-CeO2-DP accelerating CO2RR[74-75]
X.C.Sun 等[75]利用沉积的Au3+进行电化学预处理,诱导了Au-CeO2界面的活化。Au3+的还原将多余的电子引导到Au-CeO2界面,从而产生部分带有正电荷的Auδ+(Au-CeO2-DP)(见图6(b))。
Auδ+不 仅 可 以 促 进CO2活 化,还 可 以 加速*COOH 的形成,从而提高CO2还原性能。结果表明,在电位为-0.7~-1.0 V 的条件下,Au-CeO2-DP 生成CO 的FE 高达95.0%;Ag+和Au3+的加入不仅使催化剂有更大的界面面积,而且通过促进CeO2的还原,加速了*COOH 的形成。因此,可以在CeO2中掺入其他半径大、化合价高的金属,探究其对CO2RR 性能的影响。
2.3 稀土元素作为助催化剂
目前,传统的过渡金属催化剂主要通过杂原子与过渡金属形成d-d 和d-p 轨道,进行电子结构的调制,从而提高催化剂的活性[76]。稀土元素具有独特的4f 轨道,可以与过渡金属形成d-f 轨道[77],能够进一步促进电子转移,降低反应中间体的能垒。因此,在过渡金属催化剂中加入稀土元素能够提高CO2RR 催化性能。
W.Q.Liu 等[78]合成Ni-Gd-N 三元掺杂多孔炭黑电催化剂,并进行了XRD、XPS、电荷转移等分析及电化学测试。结果表明,催化剂中Ni(+1)活性位点以单原子的形态暴露在碳层外,是吸附和转化CO2的活性位点,而Ni 纳米颗粒则被包裹在碳层的内部;加入Gd 原子时,部分Gd 原子被纳入Ni 纳米粒子的晶格中,并与Ni 原子形成Gd-Ni,诱发了Ni 的晶格缺陷;加入Gd 原子后,CBNNiGd-700 的衍射峰较弱,说明Gd 原子的加入抑制了Ni 纳米粒子的团聚(见图7(a));CBNNiGd-700 相对于CBNGd-700结合能出现了正移,相对于CBNNi-700 的结合能出现了负移,表明CBNNiGd-700 中Gd(+3)原子通过镧系收缩将Ni(+1)原子最外层轨道上的部分电子拉向自身,改变了Ni 的局部电子密度,从而增强了CBNNiGd-700 的催化活性(见图7(b)及图7(c));Gd 原子的引入导致CBNNiGd-700 中的Ni 活性位点有电子损失,从而导致Ni 原子上的正电荷密度较高(见图7(d),绿色表示转出电子,红色表示转入电子);在Ni 基催化剂中引入Gd 原子,能够增强Ni对*COOH 的吸附,从而有助于稳定*COOH(见图7(e));CBNNiGd-700 生成CO 的最高FE 为97.0%,对应的j(CO)为308 mA/cm2(见图7(f))。
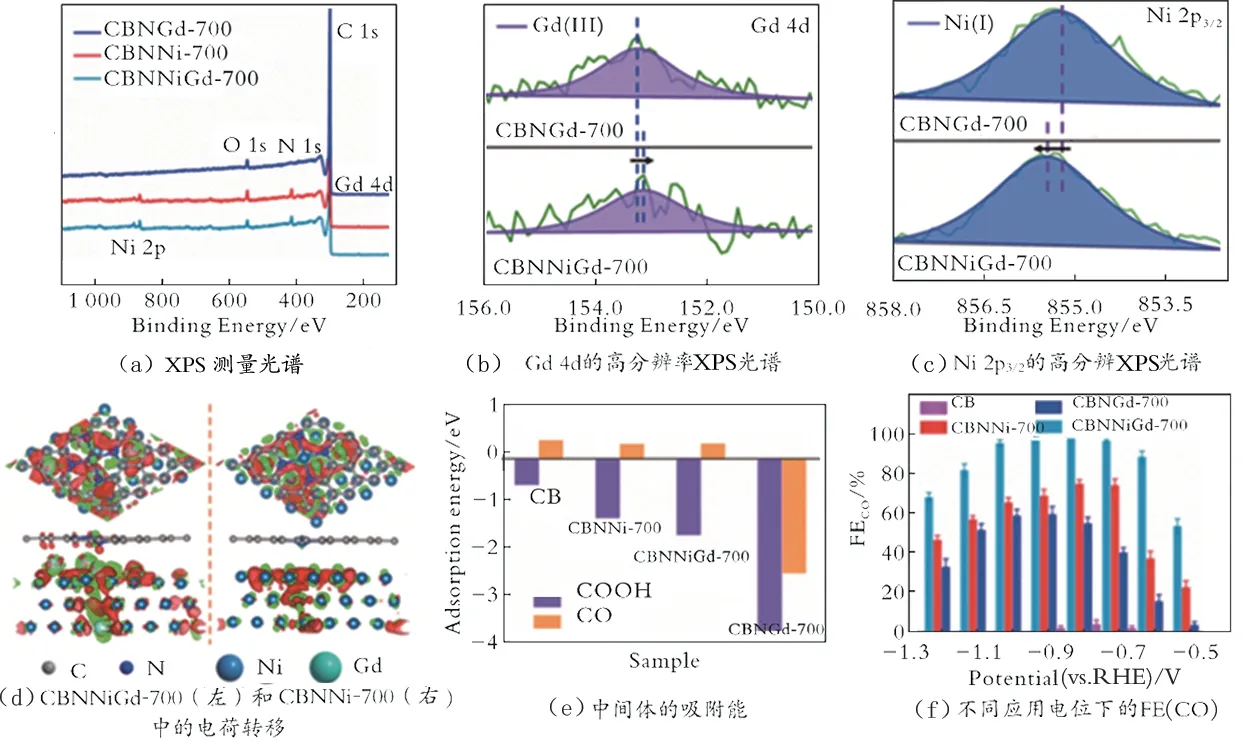
图7 CBNGd 的电子结构变化图和CO2RR 性能测试图[78]Fig.7 Electronic structure change diagram of CBNGd and CO2RR performance test diagram[78]
Z.Liang 等[79]通过调整n(Zn)/n(La),在电位范围较宽的条件下,制备了n(CO)/n(H2)范围较大的合成气,并进行了XPS 分析。结果表明,在合成气的生产过程中,Zn 促进CO2RR,而La 促进HER;相较于Zn2O3和La2O3,ZnLa-1 的Zn 2p 和La 3d 结合能位置出现了正移,表明N 改变了La 和Zn 的原子轨道,使La 和Zn 的电子结构在NC 载体上发生了重整(见图8);随着ZnLa/NC 中n(Zn)/n(La)的增加,Zn 3d 的电子结构没有发生变化,La 5d 轨道向费米能级移动,导致电子密度增加,从而促进H2的生成;由Zn 和La原子锚定引起的电子结构的调制,既保证了合成气的强电子转移,又保证了合成气的高FE。综上可知,稀土元素不仅能够与其他过渡金属元素结合,而且可利用稀土元素与过渡金属之间的协同作用,控制反应中间体的吸附和解析;还可以通过调节稀土元素与其他金属的物质的量比生产合成气;利用稀土元素调节电子结构,提高CO2RR 还原性能。

图8 ZnLa 的XPS 精细谱图和部分态密度图[79]Fig.8 XPS fine spectrum and partial density of states of ZnLa[79]
3 结论与展望
稀土元素具有独特的4f 轨道和未填充的5d 轨道,因此在CO2RR 中具有独特的作用。当稀土元素单原子作为主催化剂时,可通过配位阴离子与稀土元素的富电子轨道成键,通过电子结构之间的相互作用使反应中间体的吸附和解吸达到适宜的状态。通过NC 对稀土元素的电子结构进行调制,可使其形成稳定的M-N 结构,更容易将CO2高效地还原为CO。当稀土元素氧化物CeO2作为载体时,可提供大量的氧空位,而氧空位可作为催化剂的活性位点,因此可提高CO2RR 性能;通过CeO2与金属的界面作用,可促进晶格氧的迁移,进一步促进Ce3+形成,加强*COOH 的稳定性;氧空位的形成导致在其周围产生额外的电子,因此更容易形成Ce3+;额外的电子被作为活性位点的金属吸引,提高CO2RR 选择性。当稀土元素作为助催化剂时,可以利用镧系收缩改变周围原子的局部电子密度,优化载体的电子结构,提高吸附CO2的能力,也可以控制其还原CO2的活性位点的金属尺寸,从而控制CO2RR 的发生。
中国作为稀土元素产量第一的国家,在稀土元素的开发利用方面具有绝对的优势。稀土元素已经被广泛应用于各行各业,在催化方面已被用于析氧反应、析氢反应、氮气还原反应以及CO2还原反应。目前,过渡金属用于电催化CO2RR 的研究较为广泛,而稀土元素在电催化CO2RR 应用方面的研究较少。稀土元素用于电催化CO2RR 的种类较少,但用于光催化还原CO2的稀土元素比较多,例如Er、Nd、Eu、Yb、La 等,可从电催化的角度考虑这些稀土元素是否可用于CO2RR。此外,还可以探讨是否能通过调整稀土元素的配位阴离子、负载量、结构及其形态提高CO2还原性能;可以利用稀土元素的4f电子,调节过渡金属基催化剂的电子结构,优化催化剂的活性位点和反应中间体之间的吸附能,从而有效地提高电催化CO2的还原性能。
猜你喜欢
少儿科学周刊·儿童版(2021年22期)2021-12-11 21:27:59
少儿科学周刊·儿童版(2021年22期)2021-12-11 21:27:59
少儿科学周刊·儿童版(2021年22期)2021-12-11 06:42:32
食品安全导刊(2021年20期)2021-11-28 00:56:56
中国有色金属学报(2018年2期)2018-03-26 07:58:37
电镀与环保(2016年2期)2017-01-20 08:15:26
中南大学学报(自然科学版)(2016年2期)2017-01-19 07:37:25
现代工业经济和信息化(2016年12期)2016-05-17 05:37:52
中国资源综合利用(2016年7期)2016-02-03 03:00:13
无机化学学报(2014年3期)2014-02-28 17:30:48
