
小儿健脾颗粒质量标准建立研究
2023-09-13 01:56胡军华章晨峰王振中
中草药 2023年18期
王 鼎,王 佳,胡军华,章晨峰,王振中,林 夏*,肖 伟*
小儿健脾颗粒质量标准建立研究
王 鼎1, 2, 3,王 佳2, 3,胡军华2, 3,章晨峰2, 3,王振中1, 2, 3,林 夏2, 3*,肖 伟1, 2, 3*
1. 南京中医药大学,江苏 南京 210000 2. 江苏康缘药业股份有限公司,江苏 连云港 222001 3. 中药制药过程控制与智能制造技术全国重点实验室,江苏 连云港 222001
建立小儿健脾颗粒(Xiao’er Jianpi Granules,XJG)的定性、定量分析方法。采用TLC法,以“整体鉴别”新模式为基础,建立了一样多测(炒莱菔子、甘草及陈皮)及一板多鉴(炒莱菔子、甘草)的鉴别方法。采用HPLC法,采用Kromasil C18色谱柱(150 mm×4.6 mm,3.5 μm),以0.1%三氟乙酸-乙腈为流动相进行梯度洗脱,进样量5 μL,柱温30 ℃,体积流量1 mL/min,检测波长252 nm(甘草酸)、275 nm(甘草苷)、283 nm(芸香柚皮苷、橙皮苷)、326 nm(3,6′-二芥子酰基蔗糖、芥子碱硫氰酸盐)。建立的TLC鉴别方法,斑点清晰,能专属的鉴别处方中白术、陈皮、山药、炒莱菔子、焦山楂、甘草;建立了芥子碱硫氰酸盐、3,6′-二芥子酰基蔗糖、橙皮苷、芸香柚皮苷、甘草苷、甘草酸含量测定方法,6个成分在各自质量浓度范围内线性关系良好(≥0.999 9),精密度、稳定性及重复性良好,加样回收率为97.8%~101.6%;4批样品中芥子碱硫氰酸盐、3,6′-二芥子酰基蔗糖、橙皮苷、芸香柚皮苷、甘草苷、甘草酸的质量浓度分别为0.57~0.80、0.44~0.64、3.81~4.93、1.45~2.72、0.56~1.14、1.20~1.58 mg/mL。建立了6个药味的TLC鉴别方法,斑点清晰且专属性强。建立了HPLC同时测定的6个成分含量测定方法,建立的方法快速、准确、专属性强,为其质量标准建立提供参考依据。
小儿健脾颗粒;HPLC;TLC;一板多鉴法;质量控制;整体鉴别;一样多测;甘草酸;甘草苷;芸香柚皮苷;橙皮苷;3,6′-二芥子酰基蔗糖;芥子碱硫氰酸盐;白术;陈皮;山药;炒莱菔子;山楂;甘草
小儿健脾颗粒(Xiao’er Jianpi Granules,XJG)为江苏康缘药业股份有限公司研发的“1.1”类新药,由茯苓、白术、陈皮、山药、焦山楂、炒麦芽、炒莱菔子、甘草共8味中药经水提工艺制成的颗粒剂。方中茯苓、白术为君药,健脾利湿;陈皮、山药共为臣药,理气防君药滋补碍胃,同时增强君药健脾之力;焦山楂、炒麦芽、炒莱菔子为佐药,健脾和胃、消食化积、通腑行气;甘草和中缓急为使药。诸药合用具有健脾消食功能,主要用于儿童功能性消化不良。方中茯苓、白术、山药和麦芽均含有多糖、氨基酸类化合物[1-3],茯苓还含有三萜类成分[4],白术还含有内酯类成分[5];陈皮中主要含有黄酮、柠檬苦素及生物碱等化合物[6];山楂中主要含有黄酮类、有机酸类、三萜类化合物[7];莱菔子主要含有硫代葡萄糖苷、含硫衍生物、黄酮苷和生物碱类化合物[8-9];甘草主要含有黄酮、三萜、香豆素类化合物[10-11]。鉴于处方药味所含化学成分复杂多样,面临药味质控指标成分缺失或专属性不强等问题[12]。有必要从整体质量控制角度进行设计,建立能够全面、有效控制XJG的质量标准。
一板多鉴和一样多测的鉴别方法,在复方阿胶浆[13]和强肝胶囊[14]等复方制剂中得到了应用,即使用一块薄层板对多个药味进行的鉴别。本研究基于整体控制[15]的思路进行鉴别研究,首次建立了采用一个供试品溶液即可同时用于XJG中炒莱菔子、甘草及陈皮的TLC鉴别方法,且炒莱菔子和甘草可用一个薄层板同时鉴别;同时建立了方中君药白术、臣药山药以及佐药焦山楂的TLC鉴别方法。能够以较少的试验次数,对方中君药(白术)、臣药(山药、陈皮)、佐药(焦山楂、炒莱菔子)、使药(甘草)进行有效鉴别。
多成分含量测定方法也广泛应用于复方制剂的质量控制[16-17]。本实验首次建立了HPLC同时快速测定XJG中芥子碱、3,6′-二芥子酰基蔗糖、橙皮苷、芸香柚皮苷、甘草苷、甘草酸的定量方法。该方法采用常规十八烷基硅烷键合硅胶柱色谱和乙腈-三氟乙酸流动相体系,即可完成处方中生物碱、糖酯、黄酮苷以及三萜类成分的同时测定。建立的6个药味的TLC鉴别方法及6个指标成分的HPLC同时测定方法,可作为全面控制XJG质量的有效方法。
1 仪器与材料
1.1 仪器
Waters Arc型高效液相色谱仪,含2998PDA型检测器,美国沃特世公司;Mettler Toledo XP6型电子分析天平(百万分之一)、Mettler Toledo MS204型电子分析天平(十万分之一),瑞士梅特勒公司;Milli-Q Academic型纯水仪,美国密理博公司;TG16MW型台式高速离心机,湖南赫西仪器装备有限公司;ATS4型全自动点样仪,香港力杨企业有限公司。
1.2 试药
1.2.1 对照品 芥子碱硫氰酸盐(批号111702-202006,质量分数99.0%)、橙皮苷(批号110721-202019,质量分数95.3%)、甘草苷(批号111610-201908,质量分数95.0%)、尿囊素(批号111501-200202,质量分数100.0%)、白术内酯II(批号111976-201501,质量分数99.9%)、3,6′-二芥子酰基蔗糖(批号111848-202006,质量分数96.5%)、熊果酸(批号110724-201823,质量分数99.9%)、甘草酸铵(批号110731-202021,质量分数96.2%),均购自中国食品药品检定研究院;芸香柚皮苷(批号ST05590120/8958,质量分数99.9%),购自上海诗丹德标准技术服务有限公司。
1.2.2 对照药材 白术(批号120925-202013)、甘草(批号120904-202021),均购自中国食品药品检定研究院。
1.2.3 样品 XJG(批号210901、211101、211102、211103)、白术阴性制剂、甘草阴性制剂、炒莱菔子阴性制剂、陈皮阴性制剂、山药阴性制剂、焦山楂阴性制剂,均来源于江苏康缘药业股份有限公司。
1.3 试剂
乙腈,色谱纯,批号020901,购自迈瑞达科技有限公司;色谱甲酸,批号202674,购自赛默飞世尔科技有限公司;三氟乙酸,分析纯,批号0200718,购自美国天地公司;AB-8型大孔树脂,批号HO4M7L14173,购自上海源叶生物科技有限公司;-丙基乙二胺(primary secondary amine,PSA),批号6386696-D1,购自安捷伦科技有限公司;活性炭,批号20220111,购自国药集团化学试剂有限公司。
2 方法与结果
2.1 TLC鉴别方法的建立
2.1.1 白术 取3批XJG(批号211101、211102、211103)粉末各2 g,加水50 mL,超声20 min,于8000 r/min离心5 min,取上清液,用乙醚萃取3次,每次20 mL,水液备用,合并乙醚液,挥干溶剂,残渣加甲醇1 mL使溶解,作为供试品溶液。取白术阴性制剂,同供试品溶液制备方法同法制备白术阴性供试品溶液。取白术对照药材0.5 g,加水25 mL加热回流1 h,放冷至室温,于8000 r/min离心5 min,上清液用乙醚萃取3次,每次20 mL,合并乙醚液,蒸干,残渣加甲醇1 mL使溶解,作为对照药材溶液。另取白术内酯II对照品,加甲醇制成含白术内酯II 0.1 mg/mL的对照品溶液。照薄层色谱法(通则0502)[18]进行试验,吸取上述溶液各5~10 μL,分别点于同一硅胶G薄层板上,以石油醚(60~90 ℃)-醋酸乙酯(5∶1)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,105 ℃加热至斑点显色清晰,置于紫外灯(365 nm)下检视。供试品溶液色谱中,在与对照品色谱相应的位置上,显相同颜色的荧光斑点,且阴性无干扰,表明该方法专属性良好。结果见图1。
2.1.2 甘草、炒莱菔子 取白术供试品溶液制备过程中备用水液,加正丁醇萃取3次,每次20 mL,合并正丁醇,挥干溶剂,残渣加甲醇2 mL使溶解,加入PSA 0.2 g混合后离心,取上清液,即得甘草、炒莱菔子供试品溶液。取甘草、炒莱菔子阴性制剂,同供试品溶液制备方法同法制备甘草、炒莱菔子阴性供试品溶液。取甘草对照药材0.2 g,参照《中国药典》2020年版一部“甘草”药材标准方法[19]中的对照药材制备方法,同法制备对照药材溶液。另取甘草苷、芥子碱硫氰酸盐加甲醇分别制成含甘草苷0.5 mg/mL、芥子碱硫氰酸盐1 mg/mL的对照品溶液。照薄层色谱法(通则0502)[18]进行试验,吸取上述溶液各2~6 μL,分别点于同一硅胶G薄层板上,以醋酸乙酯-甲酸-水(10∶2∶3)的上层溶液为展开剂,展开,取出,晾干,置紫外灯(365 nm)下检视,结果供试品色谱中,在与对照品色谱及对照药材相应的位置上,显相同颜色的荧光斑点,且阴性无干扰(炒莱菔子)。再喷以10%硫酸乙醇溶液,105 ℃加热至斑点显色清晰,置紫外光灯(365 nm)下检视,结果供试品色谱中,在与对照品色谱及对照药材相应的位置上,显相同颜色的荧光斑点,且阴性无干扰(甘草)。上述结果表明方法均专属性良好。结果见图2。
2.1.3 陈皮 供试品制备方法同“2.1.2”项下。取陈皮阴性制剂,同法制备陈皮阴性供试品溶液。另取橙皮苷对照品,加甲醇制成0.4 mg/mL对照品溶液。照薄层色谱法(通则0502)[18]进行试验,吸取上述各溶液1~3 μL,分别点于同一硅胶G薄层板上,以醋酸乙酯-甲醇-水(100∶17∶13)为展开剂,展开,取出,晾干,喷以三氯化铝试液,置紫外光灯(365 nm)下检视。结果供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的荧光斑点,且阴性无干扰,表明该方法专属性良好。结果见图3。
2.1.4 山药 取3批XJG(批号211101、211102、211103)粉末5 g,加60%乙醇50 mL,加热回流30 min,趁热滤过,滤液蒸干,残渣加水5 mL使溶解,加于AB-8型大孔吸附树脂柱(内径为1.5 cm,柱高为18 cm)上,用水100 mL洗脱,收集洗脱液,洗脱液加于活性炭层析柱(内径为1.5 cm,柱高为10 cm)上,以2滴/s的速度过活性炭层析柱,用水50 mL洗脱,弃去水液,再用50%乙醇50 mL洗脱,收集洗脱液,蒸干,残渣加乙醇1 mL,置水浴上加热溶解,即得供试品溶液。取山药阴性制剂,同法制得山药阴性供试品溶液。另取尿囊素对照品,加50%甲醇制成含尿囊素0.2 mg/mL的对照品溶液。照薄层色谱法(通则0502)[18]进行试验,吸取上述溶液各5 μL,分别点于同一硅胶G薄层板上,以丙酮-三氯甲烷-醋酸乙酯-甲酸-甲醇(5∶1∶8∶1∶2)为展开剂,展开,取出,晾干,喷以15%对二甲氨基苯甲醛的10%硫酸乙醇溶液,加热至斑点显色清晰。结果供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点,且阴性无干扰,表明该方法专属性良好。结果见图4。
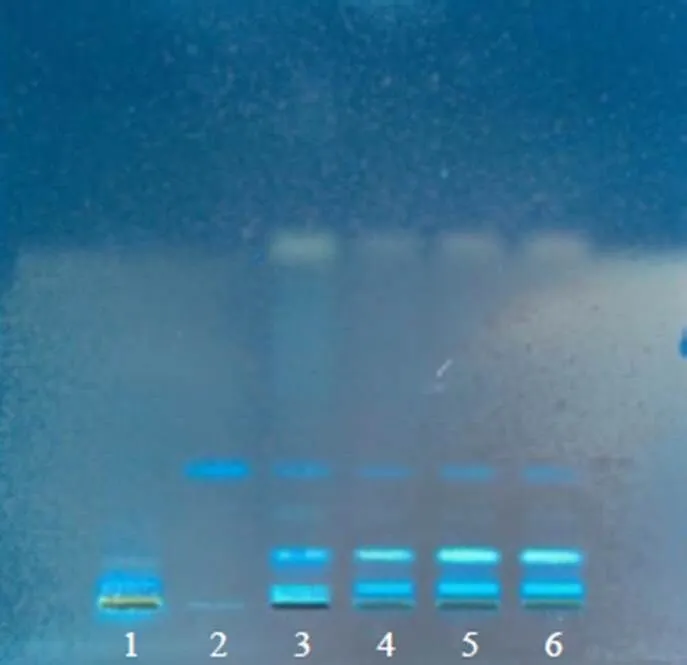
1-白术阴性供试品溶液 2-白术内酯II对照品溶液 3-白术对照药材溶液 4~6-XJG供试品溶液(批号21101、211102、211103)

1-山药阴性供试品溶液 2-尿囊素对照品溶液 3~5-XJG供试品溶液(批号211101、211102、211103)
2.1.5 焦山楂 取3批XJG(批号211101、211102、211103)粉末15 g,加二氯甲烷80 mL,加热回流2 h,滤过,取滤液,用水洗3次,每次80 mL,分取二氯甲烷液,蒸干,残渣加二氯甲烷2 mL使溶解,作为供试品溶液。取缺焦山楂阴性制剂,同供试品溶液制备方法制成焦山楂阴性对照溶液。取熊果酸对照品,加甲醇制成含熊果酸1 mg/mL的对照品溶液。照薄层色谱法(通则0502)[18]进行试验,吸取上述2种溶液各4~6 μL,分别点于同一硅胶G薄层板上,以环己烷-醋酸乙酯-甲酸溶液(20∶6∶1)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105 ℃加热至斑点显色清晰,置紫外光灯(365 nm)下检视。结果供试品色谱中,在与对照品色谱相应的位置上,显相同的橙黄色荧光斑点,且阴性无干扰,表明该方法专属性良好。结果见图5。

1-焦山楂阴性供试品溶液 2-熊果酸对照品溶液 3~5-XJG供试品溶液(批号211101、211102、211103)
2.2 HPLC定量分析
2.2.1 色谱条件 色谱柱为Kromasil C18(150 mm×4.6 mm,3.5 μm);流动相为0.1%三氟乙酸水溶液-乙腈,梯度洗脱:0~12 min,5%~19%乙腈;12~15 min,19%~21%乙腈;15~23 min,21%~26%乙腈;23~45 min,26%~90%乙腈;柱温30 ℃;体积流量1.0 mL/min;进样量5 µL;检测波长252 nm(甘草酸铵)、275nm(甘草苷)、283 nm(芸香柚皮苷、橙皮苷)、326 nm(3,6′-二芥子酰基蔗糖、芥子碱硫氰酸盐);理论塔板数均应不低于5000。
2.2.2 对照品溶液的制备 取芥子碱硫氰酸盐对照品适量,精密称定,加0.1%三氟乙酸甲醇制成含芥子碱硫氰酸盐12 µg/mL的对照品溶液;取甘草酸铵、甘草苷、橙皮苷、芸香柚皮苷、3,6′-二芥子酰基蔗糖对照品适量,精密称定,加甲醇制成含甘草酸铵20 µg/mL、甘草苷10 µg/mL、橙皮苷80 µg/mL、芸香柚皮苷40 µg/mL、3,6′-二芥子酰基蔗糖10 µg/mL的混合对照品溶液。
甘草酸质量=甘草酸铵质量/1.0207
2.2.3 供试品溶液的制备 取装量差异项下样品,研细,取约0.4 g研细样品,精密称定,置具塞锥形瓶中,精密加入80%甲醇25 mL,称定质量,超声处理(500 W、40 kHz)40 min,放冷,用80%甲醇补足减失的质量,摇匀,离心,取上清液作为供试品溶液。
2.2.4 阴性供试品溶液的制备 取陈皮阴性、甘草阴性、炒莱菔子阴性样品,按“2.2.3”项下供试品溶液制备方法,制备阴性供试品溶液。
2.2.5 专属性试验 精密吸取“2.2.2”项下对照品溶液、“2.2.3”项下供试品溶液和“2.2.4”项下阴性供试品溶液,按“2.2.1”项下色谱条件进行测定,结果见图6、7,结果表明阴性样品无干扰。
2.2.6 线性关系考察 精密称定芥子碱硫氰酸盐对照品,加0.1%三氟乙酸甲醇溶液配制成质量浓度为200.972 μg/mL的对照品储备液1;精密称定橙皮苷和甘草酸铵对照品,加甲醇制成质量浓度分别为379.999、97.085 μg/mL的混合对照品储备液2;精密称定芸香柚皮苷、甘草苷和3,6′-二芥子酰基蔗糖对照品,加甲醇制成质量浓度分别为539.860、112.765、115.260 μg/mL的混合对照品储备液3。取以上储备液作为母液逐倍稀释,分别精密吸取5 μL,注入液相色谱仪,以峰面积平均值为纵坐标(),对照品质量浓度为横坐标(),绘制回归曲线,进行线性回归,得各对照品线性范围、回归方程和相关系数(),相关结果分别为芥子碱硫氰酸盐=16 182.0-6 713.7,=0.999 9,线性范围4.019~80.389 μg/mL;3,6′-二芥子酰基蔗糖=14 225.0-11 011.0,=0.999 9,线性范围3.602~115.260 μg/mL;甘草酸=4 046.0-2 701.1,=0.999 9,线性范围4.854~97.085 μg/mL;甘草苷=9 524.1+1 772.8,=1.000 0,线性范围3.524~112.765 μg/mL;橙皮苷=9 388.07-1 161.66,=1.000 0,线性范围19.000~379.999 μg/mL;芸香柚皮苷=8 045.1+15 841.0,=1.000 0,线性范围16.871~539.860 μg/mL。
2.2.7 精密度试验 取供试品溶液(批号210901),连续进样6次,以峰面积计算各指标成分RSD,分别为芥子碱硫氰酸盐1.44%、橙皮苷0.27%、芸香柚皮苷0.20%、甘草苷0.32%、甘草酸铵0.27%、3,6′-二芥子酰基蔗糖0.42%,结果表明仪器精密度良好。
2.2.8 稳定性试验 取供试品溶液(批号210901),于制备后0、4、8、12、24、34 h注入高效液相色谱仪进行分析,以峰面积计算各指标成分RSD,分别为芥子碱硫氰酸盐0.24%、橙皮苷0.19%、芸香柚皮苷0.56%、甘草苷0.66%、甘草酸铵0.66%、3,6′-二芥子酰基蔗糖1.70%;取同一对照品溶液,质量浓度为芥子碱硫氰酸盐0.015 mg/mL、橙皮苷0.076 mg/mL、芸香柚皮苷0.067 mg/mL、甘草苷0.014 mg/mL、甘草酸铵0.019 mg/mL、3,6′-二芥子酰基蔗糖0.014 mg/mL,于制备后0、4、8、12、24、34 h注入高效液相色谱仪进行分析,以峰面积计算各指标成分RSD,分别为芥子碱硫氰酸盐0.62%、橙皮苷0.81%、芸香柚皮苷0.40%、甘草苷0.51%、甘草酸铵0.82%、3,6′-二芥子酰基蔗糖1.52%。结果RSD均小于2%,表明供试品溶液和对照品溶液在34 h内稳定性良好。

图6 溶剂(A)、阴性样品溶液(B)、混合对照品溶液(C)和XJG供试品溶液(D)在不同波长下的HPLC图

图7 溶剂(A)、阴性样品溶液(B)、芥子碱硫氰酸盐对照品溶液(C)、混合对照品溶液(D)和XJG供试品溶液(E)在326 nm下的HPLC图
2.2.9 重复性试验 取同一供试品(批号210901),按“2.2.3”项方法制备供试品溶液,平行制备6份,以本研究色谱条件进行分析,进行重复性结果验证。各成分质量分数分别为芥子碱硫氰酸盐0.79 mg/g、橙皮苷5.05 mg/g、甘草酸1.38 mg/g、3,6′-二芥子酰基蔗糖0.62 mg/g、甘草苷0.58 mg/g、芸香柚皮苷2.65 mg/g,RSD分别为0.38%、0.23%、0.61%、0.90%、0.51%、0.62%,结果表明方法重复性良好。
2.2.10 加样回收率试验 称取已测定的XJG样品(批号210901)9份,分为高、中、低3组,每组3份,各0.2 g,精密称定,分别精密加入线性关系考察“2.2.6”项下储备液(储备液1:芥子碱硫氰酸盐161.410 μg/mL,低、中、高各加0.5、1.0、2.0 mL;储备液2:橙皮苷379.999 μg/mL、甘草酸铵97.085 μg/mL,低、中、高各加1.0、2.5、4.0 mL;储备液3:芸香柚皮苷539.860 μg/mL、甘草苷112.765 μg/mL、3,6′-二芥子酰基蔗糖115.260 μg/mL,低、中、高各加0.5、1.0、1.5 mL),再精密加入80%甲醇补足至25 mL,以重复性试验各成分质量分数计算加样回收率,结果芥子碱硫氰酸盐、3,6′-二芥子酰基蔗糖、甘草酸铵、芸香柚皮苷、甘草苷、橙皮苷的平均加样回收率分别为98.7%、99.5%、101.5%、101.6%、97.8%、100.9%,RSD分别为1.5%、3.4%、1.7%、1.5%、1.1%、2.0%,结果表明该方法准确度良好。
2.2.11 4批制剂定量分析结果 取4批XJG制剂(批号210901、211101、211102、211103)各10袋,研细,分别按“2.2.3”项下方法制备供试品溶液,并按照“2.2.1”项下色谱条件进样分析,分别计算6个指标成分的含量,结果见表1。
甘草苷和芸香柚皮苷的含量批间差异较大,经分析发现主要为投料饮片含量批间差异大导致,后续可进一步制定药材、饮片内控标准,细化工艺确保批间一致性。
表1 XJG中6个主要成分含量
Table 1 Contents of six main components of XJG
成分质量分数/(mg∙g−1)含量/(mg∙袋−1) 芥子碱硫氰酸盐橙皮苷甘草酸甘草苷3,6′-二芥子酰基蔗糖芸香柚皮苷芥子碱硫氰酸盐橙皮苷甘草酸甘草苷3,6′-二芥子酰基蔗糖芸香柚皮苷 2109010.804.931.290.600.642.722.0012.33.231.461.556.63 2111010.754.981.320.560.602.261.8412.203.251.381.485.56 2111020.654.181.200.590.531.841.5810.202.951.441.294.51 2111030.573.811.581.140.441.451.409.403.892.791.083.55
3 讨论
3.1 TLC鉴别供试品溶液制备方法优选
考察了样品经水超声提取后,将水液先后采用乙醚、正丁醇萃取,对不同极性成分进行富集。取乙醚萃取得供试品溶液用于鉴别白术,结果白术鉴别阴性无干扰,专属性强;取正丁醇萃取得供试品溶液鉴别陈皮、炒莱菔子、甘草,结果甘草鉴别阴性样品中有干扰,加入适量PSA后可以有效的去除有机酸和色素,使斑点更加清晰,结果优化后的供试品制备方法陈皮、炒莱菔子、甘草鉴别阴性无干扰,专属性强。
3.2 含量测定色谱条件优选
结合处方工艺,选取制剂中含量在万分之二以上成分,芥子碱、3,6′-二芥子酰基蔗糖、橙皮苷、芸香柚皮苷、甘草苷、甘草酸作为定量指标。采用苯基柱,考察了以乙腈-3%乙酸为流动相,芥子碱硫氰酸盐分离较好,但是其他5个成分分离度较差;采用常规C18柱,考察了乙腈-0.1%甲酸水溶液、乙腈- 0.1%三氟乙酸水溶液、乙腈-3%乙酸水溶液3种流动相体系,结果以乙腈-0.1%甲酸水溶液、乙腈-3%乙酸水溶液为流动相时,其他成分分离较好,芥子碱硫氰酸盐保留差,分离度达不到要求;以乙腈-0.1%三氟乙酸水溶液为流动相时,芥子碱硫氰酸盐保留适中,峰形较好,且其他成分的分离度均在1.5以上,最终选择了乙腈-0.1%三氟乙酸水溶液作为流动相。考察了4个品牌色谱柱Kromasil C18、Waters Symmetry、Fortis H2O、Shimadzu C18-AQ,结果6个指标成分均能得到较好分离。
3.3 对照品溶液配制溶剂优选
研究发现芥子碱硫氰酸盐稳定性差。分别采用甲醇、50%甲醇、0.1%三氟乙酸甲醇溶液、3%冰醋酸甲醇溶液配制芥子碱硫氰酸盐对照品,考察芥子碱硫氰酸盐在溶液中的稳定性。结果以甲醇、50%甲醇为溶剂时,芥子碱硫氰酸盐在8 h内峰面积下降显著;以0.1%三氟乙酸的甲醇溶液、3%冰醋酸甲醇溶液作为溶剂时,在冷藏的条件下,30 d内芥子碱硫氰酸盐含量无显著变化。故选择采用0.1%的三氟乙酸甲醇溶液作为芥子碱硫氰酸盐的配制溶剂。
3.4 小结
本研究建立的6个药味的TLC薄层鉴别方法及6个成分含量测定方法可以较为全面地对XJG进行质量控制。但是鉴于本品的水提工艺,暂未能建立处方中茯苓和麦芽的专属性成分鉴别,后续有待进一步研究。含量测定指标成分3,6′-二芥子酰基蔗糖、芸香柚皮苷在处方药味中均未进行控制,后续将继续开展药材质量标准提升研究,并建立中间体质量控制标准。通过药材-中间体-制剂质控体系的建立,最终实现全过程的质量控制。
利益冲突 所有作者均声明不存在利益冲突
[1] 朱赟斐, 谭善忠, 王洪兰, 等. 基于UPLC-Q-TOF-MS/ MS技术的益气健脾颗粒化学成分分析 [J]. 中草药, 2022, 53(12): 3601-3613.
[2] Luo L, Cai J, Zhou Z,. Polysaccharides from: A review on their extraction, purification, structure, and bioactivities [J]., 2022, 2022: 2338533.
[3] 凌俊红. 麦芽的化学成分及炮制学研究 [D]. 沈阳: 沈阳药科大学, 2007.
[4] 冯群, 姚景春, 范玉兰, 等. 基于“五原则”的开心散质量标志物 (Q-Marker) 的预测分析 [J]. 中草药, 2022, 53(11): 3550-3556.
[5] Deng M, Chen H J, Long J Y,. Atractylenolides (I, II, and III): A review of their pharmacology and pharmacokinetics [J]., 2021, 44(7): 633-654.
[6] 黄芳, 周熙, 罗辉泰, 等. 基于HPLC-Q-TOF-MS及化学模式识别方法对陈皮的化学成分快速鉴别及产地判别研究 [J]. 中草药, 2022, 53(20): 6361-6368.
[7] 董嘉琪, 陈金鹏, 龚苏晓, 等. 山楂的化学成分、药理作用及质量标志物(Q-Marker)预测 [J]. 中草药, 2021, 52(9): 2801-2818.
[8] Gao L, Li H, Li B Q,. Traditional uses, phytochemistry, transformation of ingredients and pharmacology of the dried seeds ofL. (), A comprehensive review [J]., 2022, 294: 115387.
[9] 高思佳, 王计瑞, 秦伟瀚, 等. 莱菔子炮制前后HPLC特征图谱及4种成分含量变化研究 [J]. 中国中医药信息杂志, 2021, 28(5): 70-75.
[10] Li F F, Liu B, Li T,. Review of constituents and biological activities of triterpene saponins frometand its solubilization characteristics [J]., 2020, 25(17): 3904.
[11] 李葆林, 麻景梅, 田宇柔, 等. 甘草中新发现化学成分和药理作用的研究进展 [J]. 中草药, 2021, 52(8): 2438-2448.
[12] 李天娇, 包永睿, 王帅, 等. 中药质量控制与评价创新方法研究进展及应用 [J]. 中草药, 2022, 53(20): 6319-6327.
[13] 许啸, 张淹, 刘晓云, 等. 复方阿胶浆质量标准提升研究 [J]. 中草药, 2021, 52(20): 6226-6233.
[14] 付莉, 韩桂茹. 三种制剂中多味药材的同时鉴别研究 [J]. 中国药事, 2012, 26(1): 69-71.
[15] 周跃华, 路金才, 周娟, 等. 关于中药新药复方制剂“整体鉴别”新模式的思考 [J]. 中草药, 2021, 52(8): 2199-2204.
[16] 张越, 陈健, 李洋, 等. 经典名方温经汤标准汤剂HPLC指纹图谱建立及9种成分含量测定 [J]. 中草药, 2020, 51(18): 4664-4672.
[17] 李华, 刘梓晗, 孟欣, 等. 经典名方清燥救肺汤的多成分含量测定方法和抗氧化活性研究 [J]. 药学学报, 2021, 56(12): 3511-3517.
[18] 中国药典[S]. 四部. 2020: 59.
[19] 中国药典[S]. 一部. 2020: 88.
Establishment of quality standard of Xiao’er Jianpi Granules
WANG Ding1, 2, 3, WANG Jia2, 3, HU Jun-hua2, 3, ZHANG Chen-feng2, 3, WANG Zhen-zhong1, 2, 3, LIN Xia2, 3, XIAO Wei1, 2, 3
1. Nanjing University of Chinese Medicine, Nanjing 210000, China 2. Jangsu Kanion Pharmaceutical Co., Ltd., Lianyungang 222001, China 3. National Key Laboratory on Technologies for Chinese Medicine Pharmaceutical Process Control and Intelligent Manufacture, Lianyungang 222001, China
To establish a qualitative and quantitative analysis method for Xiao’er Jianpi Granules (小儿健脾颗粒, XJG).Based on the new pattern of “holistic identification”, TLC was used to establish the identification methods of one-sample multi-detection [stir-fried Laifuzi (, sRS), Gancao (et, GRR) and Chenpi (, CRP)] and one-plate multi-detection (sRS, GRR). HPLC was performed on a Kromasil C18column (150 mm × 4.6 mm, 3.5 μm) with a gradient elution using 0.1% trifluoroacetic acid-acetonitrile as the mobile phase, the injection volume was 5 μL, the column temperature was 30 ℃, the flow rate was 1 mL/min, and the detection wavelengths were 252 nm (glycyrrhizic acid), 275 nm (liquiritin), 283 nm (narirutin, hesperidin), 326 nm (3,6′-disinapoyl sucrose, sinapine thiocyanate).The established TLC identification methods with clear spots, could exclusively identify Baizhu (, AMR), CRP, Shanyao (, DR), sRS, Shanzha (, CF) and GRR in the prescription. A method was established for the determination of sinapine thiocyanate, 3,6′-dierosyl sucrose, hesperidin, narirutin, liquiritin and glycyrrhizic acid, the linear relationship of the six main components in their respective concentration ranges was good (≥ 0.999 9) and the precision, stability and repeatability were good. The recoveries were 97.8%—101.6%. The mass concentrations of sinapine thiocyanate, 3,6′-dierosyl sucrose, hesperidin, narirutin, liquiritin and glycyrrhizic acid in four batches of sample were 0.57—0.80, 0.44—0.64, 3.81—4.93, 1.45—2.72, 0.56—1.14, 1.20—1.58 mg/mL, respectively.The TLC method for the identification of six medicinal flavors was established, and the identification was clear and exclusive. The HPLC method for the simultaneous determination of six components was established. The established method is rapid, accurate and specific, which provides a reference basis for the establishment of quality standards.
Xiao’er Jianpi Granules; HPLC; TLC; one-plate multi-detection; quality control; holistic identification; one-sample multi-detection; glycyrrhizinate; liquiritin; narirutin; hesperidin; 3,6′-disinapoyl sucrose; sinapine thiocyanate;;;; stir-fried;;et
R283.6
A
0253 - 2670(2023)18 - 5933 - 08
10.7501/j.issn.0253-2670.2023.18.011
2023-02-11
国家中医药管理局—基于重点研究室研究领域的中医药多学科研究能力提升项目:中药提取精制新技术
王 鼎,男,硕士研究生,研究方向为中药制药技术与产品开发。E-mail: 1162862224@qq.com
肖 伟,中国工程院院士,研究员级高级工程师,博士生导师,研究方向为中药新药的研究与开发。E-mail: kanionlunwen@163.com
林 夏,女,硕士,高级工程师/副主任中药师,研究方向为中药新药研发。Tel: 15105130710 E-mail: linxia297125856@163.com
[责任编辑 郑礼胜]
猜你喜欢
中国民间疗法(2021年17期)2021-11-04
生物技术通报(2021年2期)2021-04-26
理化检验-化学分册(2020年5期)2020-06-15
农药科学与管理(2019年8期)2019-11-23
故事会(2019年1期)2019-01-11
新世纪智能(语文备考)(2018年11期)2018-12-29
宝藏(2017年2期)2017-03-20
东方艺术·国画(2015年3期)2015-08-20
妇女生活(2015年7期)2015-07-20
妇女生活(2015年7期)2015-07-20
