
Extra contribution to the crystal stability of insensitive explosive TATB: The cooperativity of intermolecular interactions
2023-07-31 13:30ZhixingZhngYitoSiToYuWeipengLiYidingMochngLiuYingzheLiuBozhouWng
Defence Technology 2023年7期
Zhi-xing Zhng ,Yi-to Si ,To Yu ,Wei-peng Li ,Yi-ding M ,Mo-chng Liu ,Ying-zhe Liu ,*,Bo-zhou Wng ,**
a State Key Laboratory of Fluorine & Nitrogen Chemicals, Xi'an Modern Chemistry Research Institute, Xi'an, 710065, PR China
b International Research Center for Renewable Energy, State Key Laboratory of Multiphase Flow in Power Engineering, Xi'an Jiaotong University, Xi'an,710049, PR China
Keywords:Cooperativity Non-covalent interaction Low-sensitivity Explosives Charge transfer
ABSTRACT An in-depth analysis on the cooperativity of intermolecular interactions including hydrogen bonding and π-π stacking in 1,3,5-triamino-2,4,6-trinitrobenzene (TATB) crystal was studied.Two quantities,cooperativity rate and energy,were defined to evaluate the nature and strength of cooperativity in a series of clusters diverging from 1D to 3D prototypes.The origin and mechanism of the cooperative effect were settled to demonstrate that the nature of cooperativity is determined by whether the non-covalent interactions compete or promote with each other,which is manifested by the changing trend of electron transfer.There exists obvious cooperative effect in intra-layer and inter-layer structures as they own the equivalent non-covalent interactions,while anti-cooperative effect is also observed if two interactions correlate with each other.On the whole,in the process of crystal formation,the apparent cooperativity is the check and balance of the two effects,which is capable to support a global interaction among all of molecules and contribute to the stabilization of system.Based on the results,one may get a new insight to understand the relationship between non-covalent interactions and low impact sensitivity.
1.Introduction
In the last decades,the development of low-sensitive or insensitive explosives have become an important subject in the research field of energetic materials [1-7].There is great demand for this kind of explosives in the regard of military equipment and civil blasting because of their excellent properties such as safety and stability at extreme environment,low sensitivity and elimination to abnormal initiation and explosion.1,3,5 Triamino-2,4,6-trinitrobenzene (TATB) is one of the typical representatives as it shows remarkable stability under high temperatures,shock,and impact [8-11].The TATB crystal has a layered structure that is composed of nearly planar layers,in which one molecule connects with the other six molecules via the intermolecular hydrogen bonds between nitro and amino groups.The adjacent molecules coming from two adjacent layers can interact with each other through the interlayer close contacts.
Plenty of researches have been devoted to unravel the nature of TATB crystal and the unique properties,especially for the low impact sensitivity [11-14].It is inferred that the key types of intermolecular interactions,hydrogen-bonding and π-π stacking make great contribution.Zhang and coworkers have proposed systematical researches on the influence of hydrogen-bonding aided π-π stacking to the low impact sensitivity [15-20],as shown in Fig.1(a).In the formation of hydrogen bonds,the relevant groups such as amino and nitro are constrained in a manner.It should limit the stretching and flipping of chemical bond that are consistent with the decomposition of TATB molecule.A strong intramolecular hydrogen bond not only contributes to the stabilization of molecules with effective π-π stacking and high packing coefficients,but also counteracts the effect of external stimuli on molecular decomposition through reversible H transfer reaction to absorb and release stimulating energy,so as to further reduce the impact sensitivity.On the other hand,the intermolecular hydrogen bonds make up of a global network connecting all molecules to buffer against external mechanical effect via the inter-layer slipping(Fig.1(b)).
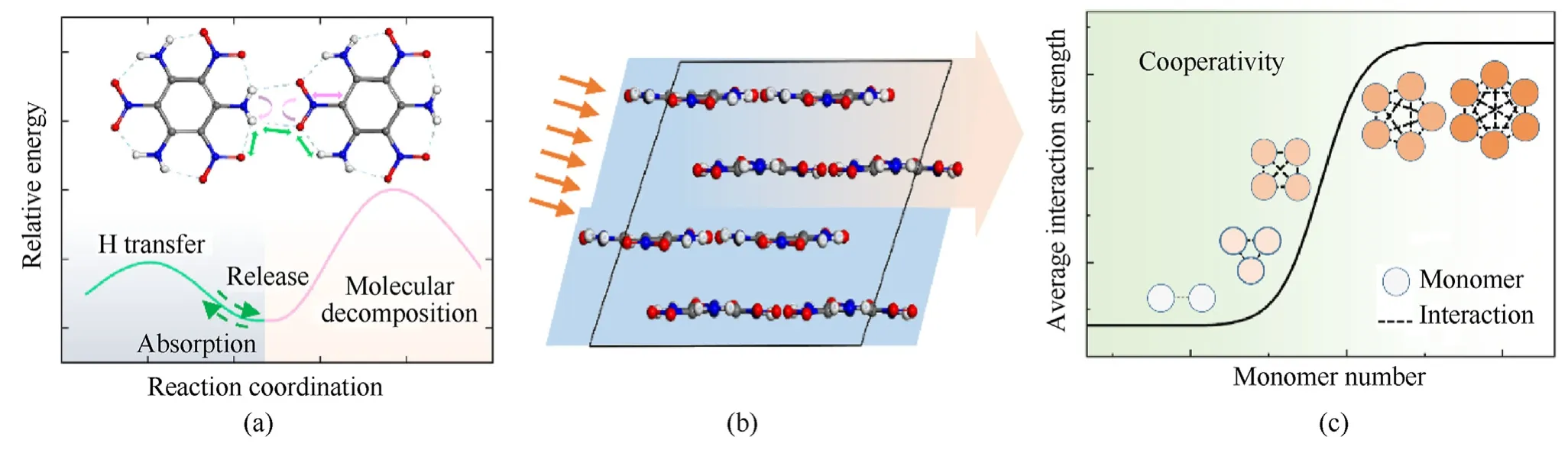
Fig.1.(a)The diagram of inhibition effect of hydrogen bond on molecular decomposition;(b)Schematic diagram of slip between molecular layers in TATB crystal;(c)Manifestation of cooperativity in TATB linear clusters.
Despite of the effects proved above,another advantage of the non-covalent interactions may also play important role in the formation and stabilization of TATB crystal,which is known as cooperativity (Fig.1(c)).The non-covalent interactions,such as hydrogen bonding,π-π stacking,halogen bonding,cation-π,anionπ,etc.[21-28],are generally treated as weak interactions.However,they may cooperate with each other to form a global effect[29,30].This type of cooperativity widely exists in the protein folding,molecular recognition,crystal formation,and so on[31-33].The total strength is usually not equal to the cumulative sum of the strengths of each non-covalent interaction but enhanced(cooperative) or weakened (anti-cooperative).
Recently,Chen and coworkers have predicted the Solid-state properties of several energetic cocrystals and demonstrated the cooperativity between hydrogen bonding and molecular polarizability [34].The cooperative effect significantly promotes the stability of the multicomponent crystals where the binding energy was increased by 19-41%,and reduces the sensitivity to external stimuli compared to their pure crystal.
Considering the work of Chen and coworkers and the typical hydrogen-bonding and π-π stacking in TATB crystal,we believe the regulating effect of cooperativity to the non-covalent interactions should not be neglected.Therefore,it is necessary to explore the existence and mechanism of cooperativity in TATB crystal.Our team performed a first principles investigation and Hirshfeld surface analysis on the crystal structure of TATB[35].Herein,we explored the cooperativity among the non-covalent interactions along with its origin and mechanism,and further tried to get a new insight to understand the relationship between non-covalent interactions and lowly mechanical sensitivity.
2.Computational details
In this work,first principles density functional theory (DFT)method was utilized for a partial optimization on the experimental crystal structure of TATB[9].All the atoms were fixed except for Hs,so as to eliminate the minor structural defect originated from the incorrect displacement of the hydrogen atoms.Perdew-Burke-Ernzerhof (PBE) functional [36] and projected augmented wave(PAW)potentials[37,38]were selected for the partial optimization.Dispersion correction was employed as an add-on (DFT-D3) [39].Considering the valence electrons of all atoms,the cut-off energy of the plane wave basis set was set as 550 eV and thek-point set was chosen as 5 × 5 × 7.The calculations are performed using the Vienna ab initio simulation package(VASP 5.4.4) [40].
In order to clarify the types of interactions in TATB crystal,plenty of clusters including different number of TATB molecules were extracted from the optimized TATB crystal structure.The extraction of molecular structures is performed in different dimensions,as shown in Fig.2.In one-dimension,the structures are formed by repeating molecules in a direction.By alternative arrangement,plenty of 1D-linears will be incorporated into the two-dimension layer structure (2D-layer) in which one molecule connects with the other six molecules and their relationships can be distinguished as three directions based on the position of hydrogen bond.Similarly,the three-dimension cluster(3D-cluster)is accumulated by 2D-layers,where the π-π stacking plays the significant role.
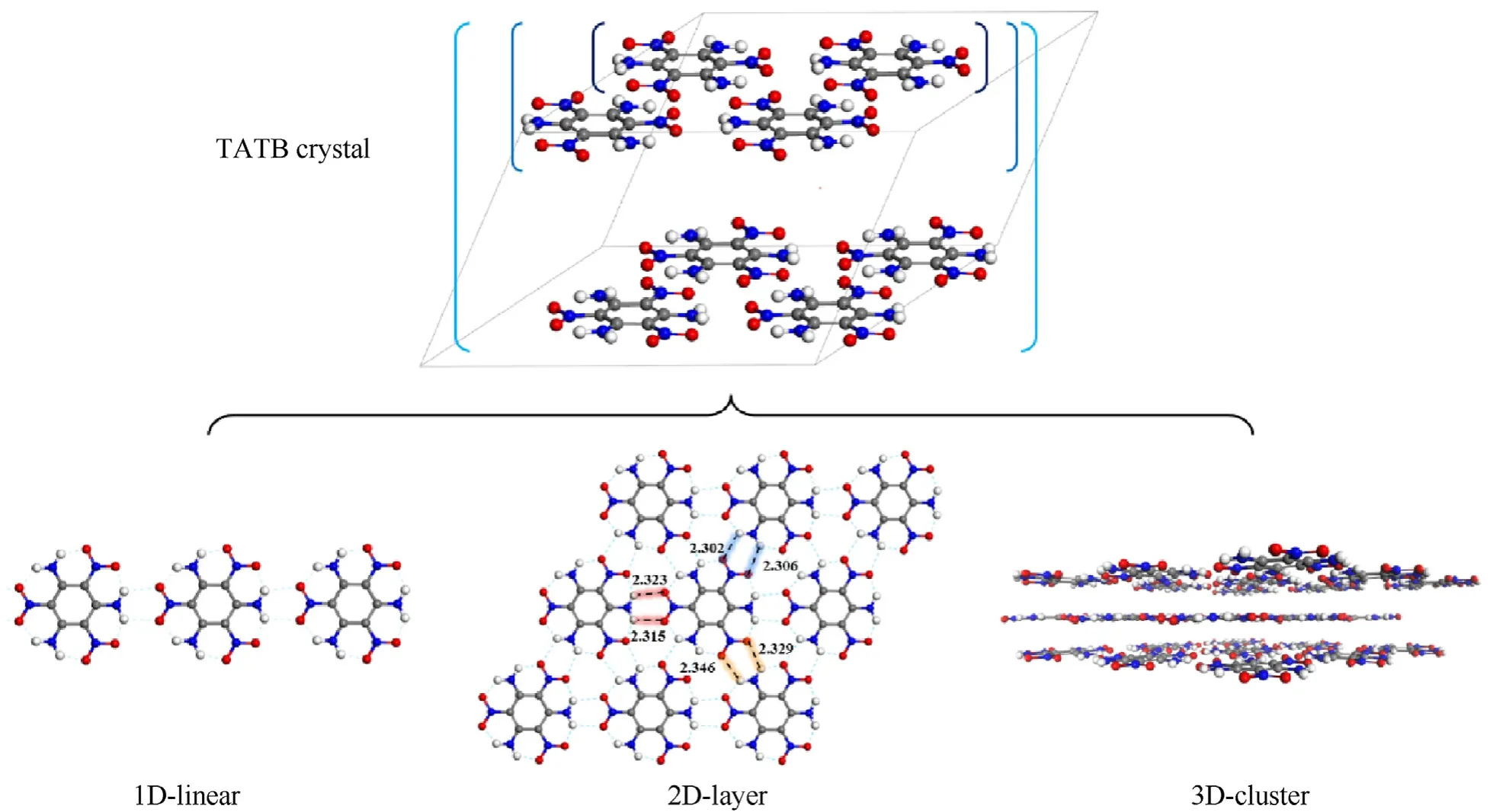
Fig.2.The diagram of extraction for TATB clusters in different dimensions.
BLYP functional [41-43] was selected to perform single-point calculations on these systems.Likewise,the atom-pairwise dispersion correction (D3BJ) was used.For the accuracy of singlepoint energy,the high-precision basis set def2-QZVPP [44] was selected.And the BSSE basis set correction was also taken into consideration [45-47].All single-point calculations were carried out under the ORCA4.2.1 software [48,49].Symmetry-Adapted Perturbation Theory (SAPT) method was adopted to make an interaction energy decomposition analysis [50].In order to study the source of the cooperative effect,we used Atomic Dipole moment Corrected Hirshfeld population (ADCH) [51] charges and dipole moment to discuss the changes in the charge distribution of each fragment of the system.The weak interaction was visualized by means of Independent Gradient Model (IGM) [52] method.Meanwhile,the electronic density difference diagram was applied to describe the electron transfer pattern in the formation of crystal.These tasks were all performed using Multiwfn 3.8.1 program[53].
In order to evaluate the cooperativity effect,three methods were employed for different conditions.Method 1 is suitable for application in the same type of interaction [54,55].For example,TATB molecules in the same layer are connected via similar hydrogenbonds.The average interaction energy ΔEintis calculated by the following Eq.(1)
wherenis the molecule number.Nintis the number of direct interactions between adjacent molecules in a layer system.Each interaction is originated from the hydrogen bonding between amino and nitro which does not include the π-π stacking or more distant neighbors,as shown in Fig.S1(a).For example,in dimer,Nintis 1.When three molecules interact with each other to form an equilateral triangle shape system,Nintchanges into 3.Enand Eiis the single point energy of the system and every molecule,respectively.According to Eq.(1),the cooperativity percentage C1 can be expressed as
where ΔEint(2) stands for the interaction energy between two molecules in the dimer which is the simplest cluster.The value and sign of C1 can give a straightforward description about the strength and nature of cooperativity.
In method 2,another cooperativity percentageC2 is defined.Different fromC1 that focuses on the global average,C2 is for individual entity.For example,any system can be divided into two random fragments,I and II.The interaction energy between them is ΔEint(old).With the extra molecules added in fragment II,the interaction energy may change into ΔEint(new).The C2 is expressed as
In TATB crystal,the molecule in any position can be treated as equivalent.Therefore,in this manuscript,a unit molecule is selected as fragment I.The other molecules left are fragment II.C2 is applied to evaluate the cooperativity relationship between the unit molecule and the other ones surrounding it,which will make an excellent description on the influence of unit molecule in crystal.
In method 3,the cooperativity energy δEis defined [56].The system studied is divided into three fragments I,II,and III.The total interaction energy is expressed as
whereE(total),E(I),E(II),E(III) are the energy of the total system and the three fragments,respectively.In the three entities system,the interaction of any two fragments should be affected by the third one.So δE is defined as
where ΔEint(I,II),ΔEint(II,III),ΔEint(I,III)are the individual pairwise interaction energies of the two fragments without the influence of third one.Compared with C1 and C2,δE owns more explicit physical meaning.If δE is negative,it means the there is a cooperativity among the fragments,and vice versa.Method 3 can be applied to the system including more fragments.But the computational complexity cannot be afforded.To simplify discussion,the systems are all divided in three fragments in this work.
3.Results and analysis
In the following discussion,several 1D,2D,and 3D systems including various number of molecules have been designed to make a detailed analysis and comparison of the cooperativity among different non-covalent interactions.
3.1.1D
As depicted in Fig.2,in layer,one molecule connects with the other six molecules via the intermolecular hydrogen bonds between nitro and amino groups.According to the hydrogen bond lengths,they can be divided into three directions,2.323/2.315 Å,2.302/2.306 Å,2.329/2.346 Å of which the interaction energies are calculated to be -4.842 kcal/mol,-4.845 kcal/mol,and -4.835 kcal/mol,respectively.On the other hand,Fig.S2 and Table S1 lists the selected bond lengths,intramolecular hydrogen bond distances,and torsional angles for TATB molecule.It can be seen that all of the C-N,N-H,and N-O bonds own high similarity.And the nitro and amino groups of TATB molecule are almost coplanar with the benzene ring.Since the differences are minor,we select the direction where the hydrogen bond lengths are 2.323/2.315 Å as standard to construct a couple of linear clusters including 2-7 molecules to have an analysis of the 1D systems.The structure diagrams are shown in Fig.3.For each system,they are divided into three fragments,black(I,first molecule),red(II,second molecule),and blue (III,molecules left).The cooperativity percentages and cooperative energy δE are listed in Table 1.

Table 1 The cooperativity percentage C1, C2,and cooperative energy δE (kcal/mol) of 1D-3~1D-7.

Fig.3.The structure diagram of linear molecular clusters 1D-2~1D-7 and the interaction energies (kcal/mol) between each two molecules.Each system is divided into three fragments,black (I,first molecule),red (II,second molecule),and blue (III,molecules left).
From 1D-2 to 1D-7,all theC1,C2,and the absolute value of δEincrease with the number of molecules,showing evident cooperative effect dominated by hydrogen bonds.The interaction energies between each two molecules have also been calculated (Fig.3),which exhibit similar tendency to cooperative percentages.To be more specific,the values between two adjacent molecules show symmetrical distribution,and the ones at center are larger than the ones outside.By comparing the interaction energies between fragment I and II,it is found that the rising tendency decreases with the number of molecules in the linear structures,which will be close to zero after a critical distance.This means the cooperativity strength is closely linked with the number of molecules in the system for 1D linear structure.
To figure out the phenomenon,the electrostatic potential(ESP)of TATB molecule was analyzed,as plotted in Fig.4(a).The ESP maximum points mainly include three kinds.First one is located on the outside of H atoms of amino group as the global maximum of 33.88 kcal/mol.The other two kinds are located on the top of sixmember rings,namely the benzene and the O-N-C-C-N-H formed by adjacent amino and nitro groups.They represent the local maxima that originate from the repulsion among the six atoms,which will play an important role in the π-π stacking of layers.Similarly,the ESP minimum points can be divided into three kinds.However,most of them are located on the top of molecular surface as local minima with positive sign,manifesting the inadequate concentration of electrons there.Only the ones located on the outside of O atoms of nitro group are negative as -26.62 kcal/mol because of concentrated electrons.Considering the location of extreme points,it is inferred that there is a strong electrostatic attraction between amino and nitro,which should make a great contribution to the hydrogen-bonding interaction.
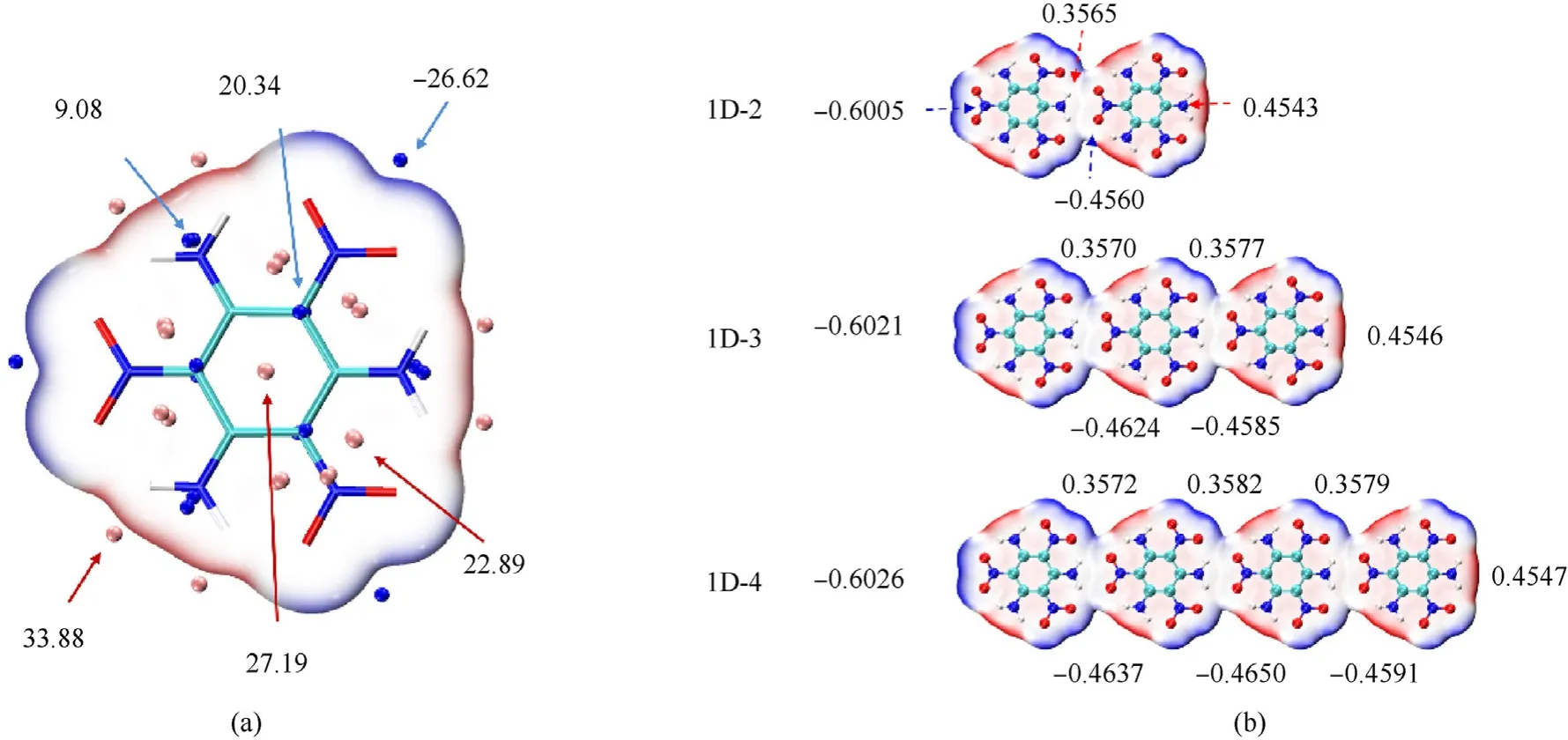
Fig.4.(a)Color-filled molecular surface map of ESP with significant surface local minima and maxima represented as blue and pink spheres,respectively.The unit is in kcal/mol.(b)The ADCH charge distributions of O atoms (blue) and H atoms (red) forming hydrogen bonds in 1D-2~1D-4.
Fig.4(b)and S3 depict the charge distributions of O atoms(blue)and H atoms(red)forming hydrogen bonds,which locate in proper negative and positive range,respectively.With the increase ofmolecule number,the absolute values both become larger,which means the polarization of the whole system becomes more obvious.In Fig.S4,1D-2 was selected as example to calculate the electron density difference diagram to visually understand the charge delocalization in the 1D-chains.In the formation of hydrogen bond,electron transfers from the H atoms to O atoms,along with a minor redistribution on C-N bond.It manifests that there exist both intermolecular and intramolecular charge transfer when the amino and nitro interact with each other.Another proof comes from the ADCH charge distribution of each molecule in 1D-2~1D-7,as listed in Table 2.In 1D-2 the first and second molecule are negatively and positively charged,respectively.It is attributed to the intermolecular charge transfer which originates from the induced polarization when the two molecules interact with each other.In 1D-3,when the third molecule is interacted with the second one,similar intermolecular charge transfer makes the former positively charged and the latter back to almost neutral.The same situation has also been observed in the other systems.And the amount of charge transfer increases as the increase of molecule number until it reaches 5.Hence,it is deduced that intermolecular and intramolecular charge transfer happen whenever a molecule is added in the linear system,which makes both the global and regional polarity of the system strengthen and promotes the electrostatic attraction between molecules [30,56,57].

Table 2 The ADCH charge(a.u.) distribution of each molecule in 1D-2~1D-7.
To assess the nature of the hydrogen bond interaction of nitro with amino group in detail,an energy decomposition analysis was performed for 1D-2 and 1D-3.The contributions of electrostatic,exchange,dispersion,and induction on the interaction energy between fragment I and the other fragments are listed in Table S2.Accordingly,most contribution to the total interaction energy is from electrostatic term,which is in agreement with the essence of hydrogen bond.The other terms should not be neglected.From 1D-2 to 1D-3,the exchange and dispersion terms almost keep constant but the electrostatic and induction terms increase,both of which are correlated with the electronic distribution.Their increase manifests that a redistribution of electrons happens in the formation of new interaction and brings the cooperativity.
According to the results above,it is concluded that every TATB molecule acts as both hydrogen-bonding donor and acceptor in the process of the linear chain formation.The hydrogen-bonding interaction is strengthened with the increase of molecule number,implying that there exists an obvious cooperativity in 1D TATB linear system.Meanwhile,the influence region of cooperative effect is limited.For a single molecule,the effect is from the other four molecules in the front and back.Analogically,in the linear system every molecule can be likened to the climber in a climbing team.And the intermolecular hydrogen bonds correlate with each other to form the rope that ties the climbers to protect them from danger.With more climbers in the team,the protection will be stronger.This is the significance of cooperativity.The thermal decomposition of TATB was studied using quantum based molecular dynamics method by He and coworkers [58].Their results proved that TATB decompositions were initiated by the homolytic cleavage of the C-NO2bond and the dehydrogenation of amino group.In other word,they are the trigger groups.Since the hydrogen-bonding can stabilize both of them,it will obstruct the decomposition of molecule to some extent.More importantly,with stronger cooperative effect among the hydrogen bonds,the obstruction will also be strengthened.
3.2.2D
Different from 1D clusters,the molecules in a 2D plain interact with each other via multiply patterns and the complexity of cooperativity grows exponentially.Herein,we select plenty of clusters with molecular number ranging from 3 to 15 to systematically explore the cooperative effect,as shown in Fig.5.

Fig.5.The structure diagrams of 2D-3a~2D-15a.
The results of C2 and δEfor these systems are depicted in Fig.6 and Table S3.Despite of their physical meaning,the two values show well quantitative correlation.Three molecules can make up two kinds of systems.For 2D-3a,the molecules interact with each other by offering ortho-position of nitro and amino,which induces a strong cooperative effect.In 2D-3b,the middle molecule interacts with the other two through the meta-position of nitro,which makes the effect turn into anti-cooperative.Combined with results of 1D-3,it is further demonstrated that if the molecule can act as both hydrogen-bonding donor and acceptor,it would bring cooperative effect to the whole system.By contrast,if the molecule only acts as either donor or acceptor,both interactions will be weakened by the competition of electron transfer,which leads to the anticooperativity.According to the unique structure of TATB,a rule can be inferred that the meta-position means competitive and anticooperative,while the ortho-/para-position means promotive and cooperative.This rule would also be applicative in other systems.

Fig.6.(a)-(f) The changing trend charts of C2 and δE in different series of systems;(g) The diagram of cooperative(yellow) and anti-cooperative(blue) effect of the surrounding molecules on the central one (orange).
When molecule number comes to four,the situation turns into more versatile.For example,2D-4a,2D-4b,and 2D-4c are designed based on 2D-3a(Fig.6(a)).The position of the forth molecule makes great effect on the strength of cooperativity.Compared with 2D-3a,2D-4a owns lower C2 and larger δEbecause the forth molecule is in competition with the fragment I but its influence to the global system is promotive.On the other hand,the smaller of δEin 2D-4b and 2D-4c is attributed to average of cooperative effect that is originated from the closer interaction with more hydrogen bonds.Similar phenomenon also happens in the comparison of 2D-3b,2D-4d,2D-4e,and 2D-4f(Fig.6(b)).It is noteworthy that 2D-4f could be treated as the combination of 1D-3 and 2D-3b.And the decrease(increase)of C2 for 2D-4d(2D-4e)manifests that there is check and balance between the competition and promotion.All of these phenomena are in consistent with the change of charge distribution.
With the molecule number increasing to five,the cooperativity mechanism becomes more obvious.2D-5a,2D-5b,and 2D-5c are designed on the basis of 1D-4(Fig.6(c)).By comparing their C2s,it is found that the influence of the fifth molecule to the cooperativity weakens as the increase of distance from fragment I,which is in agreement with the conclusion obtained in last section.In Fig.6(d),based on 1D-3,the rest 2D-5s are divided into two series,2D-5d,2D-5e,2D-5f (dash lines) and 2D-5g,2D-5h,2D-5i (dot lines).The first series shows a tendency from cooperative to anti-cooperative.While in the other series the enhancement of cooperativity is observed.The reason is that the extra two molecules interact with fragment I and II in a tighter way which induces much stronger cooperative effect.This comparison implies that the judgement of cooperativity to a system is highly dependent on the way of treatment.More discusses will be mentioned in next section.One more worth noting is that 2D-5f owns the largest C2 among 2D-5s,which is reasonable as all the other molecules that make cooperative contribution to fragment I and their effects are superposed.By contrast,in 2D-6a the sixth molecule makes an anti-cooperative effect on fragment I,which accounts for its C2 reduced.The same results also appear in the comparison of 2D-7a and 2D-9a.Moreover,the δEis decreased by the average effect originated from the enlargement of system.
To have a more detailed understanding about the rule of cooperativity contributed by the tight interaction and the enlargement of system,two series are selected.The first series is 1D-2,2D-3a,2D-4c,2D-5d*,2D-6b,and 2D-7b (Fig.6(e)).It seems that with more molecules around central fragment I the cooperativity becomes stronger,which depicts an undulating growth trend.From 1D-2 to 2D-7b both the cooperativity and anti-cooperativity strength increase.The final effect remains settled on their balance.The second series is 1D-3,2D-5i,2D-7a,2D-9b,2D-11a,and 2D-13a (Fig.6(f)).In this series,the undulating growth trend of cooperativity becomes more obvious.Combined with the results of first series,one may realize the great anti-cooperative and cooperative effect of the outer molecule on the inner and central molecule,respectively.
The C1 of these clusters have also been listed in Table S3 and Fig.S5 to analyze the cooperativity of 2D systems in global perspectives,which shows another change trend.In most cases the values of C1 are positive,which means that in the layer the global interaction among molecules should be assigned as cooperative.The exception of 2D-3b (-2.66%) and 2D-4d (-3.14%) is because there are only meta-position interactions in the two clusters.Compared with 2D-3b,the position of the forth molecule in 2D-4d bring extra anti-cooperative effect,which results in the more negative of C1 (Fig.S5(a)).On the other hand,from 2D-4e (0.39%),2D-4f(4.29%),to 2D-4b(4.73%),the effect changes into cooperative merely by shifting the forth molecule.More importantly,the trend implies if the molecules connect with each other more closely,C1 will becomes larger.Likewise,with the increase of molecule number,the situation becomes complicated.But the conclusion is proved to be settled.For example,2D-5d owns the largest C1 in 2D-5s.
In 1D clusters,we have proved that C1 will converge to constant when the cluster size is large enough.To figure out whether there exists similar phenomenon in 2D clusters,two series have been selected.One series includes 2D-3a,2D-4c,2D-5d*,2D-6b,and 2D-7b (Fig.S5(b)).With more molecules around the central one,C1 changes in an undulating trend until it reaches 4.84%.The other series includes 2D-3a,2D-6a,2D-10a,and 2D-15a (Fig.S5(c)) in which the molecules are arranged as a triangular square matrix.In this case,C1 still increase with the size of cluster but the trend becomes smaller so that it reaches to a constant value in 5.90%.Although more larger clusters containing 21 or 28 molecules are not calculated,it is believed C1 will converge into a constant when the cluster size is large enough.And its value is correlated with the structure of cluster.
In summary,through an in-depth analysis of various systems,a complete description of the cooperativity in 2D has be obtained,as shown in Fig.6(g).The yellow and blue ones surrounding make cooperative and anti-cooperative effect on central molecule,respectively.Although the effect weakens along with the increase of the distance,it has been strong enough to support a global interaction among all of molecules in a layer,which forms the ideal apparent cooperativity that contributes more than 10% to the stability of the whole layer.Regardless of the complex case,the essential mechanism is the same as in 1D and it can be treated as an enhanced version.Hydrogen bonds restrain the twist of amino and nitro groups in a more complete way to keep the molecules in a plain,which is advantageous to π-π stacking,i.e.,will favor low impact sensitivity.In this process,the cooperative effect offers assistance.It represents the uniting of all the intralayer interactions,which will help to sustain the layer integrity.
3.3.3D
In 3D,the introduction of other layers brings extra π-π interaction that makes cooperativity more complicated.Fig.7 depicts the structure of clusters with two layers.With the bottom layer asbackground,the molecules on the top layer can be arranged in three directions,blue,red,and yellow.It is obvious that in the red direction the parallel overlapping between two layers is the highest,which is more like parallel dislocated in the other two.Another worth noting is that almost every heavy atom (C,N,O) in the top layer can be projected onto the center of a six-membered ring in the bottom layer,which is similar to the phenomenon in graphite that simultaneously enhances the electrostatic attraction and avoids the collision of between π electrons.Accordingly,three systems are designed on each direction named 3D-1,3D-2,and 3D-3.As there are different non-covalent interactions,only method 3 is used to evaluate the cooperativity.The systems are divided into three fragments,the two molecules on the top layer and the molecules on bottom layer.Corresponding δEs have been calculated and listed in Table 3.3D-2 owns a positive δE,proving there is an obvious anticooperativity among the three fragments.However,in 3D-1 and 3D-3 a negligible cooperativity is observed.
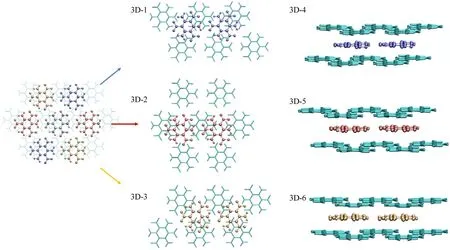
Fig.7.The structure diagrams of 3D-1~3D-6.
To figure out the reason,their inter-IGM and electronic density difference diagrams are compared,as shown in Fig.8.The most obvious interactions between two layers are localized on the position which is between the O atoms on a special nitro (top layer)and the center of benzene(bottom layer).Interestingly,the special nitro is just the hydrogen-bonding acceptor in 3D-2.It is speculated that the formation of interaction between layers will lead to a charge redistribution on the relevant atoms or substitutes.The speculation is proved in the electronic density difference diagram.The localization of pink isosurfaces on the heavy atoms indicates the electron transfer pattern when two layers are close to each other,which shows that electrons are attracted from inside of layer to outside of layer.The attraction influences the electron distribution of the nitro and amino forming hydrogen bond and reduces the electrostatic interaction between them,which will make the noncovalent interaction weaken.Therefore,the cooperativity in 3D-2 should be negative.The reason it changes into positive and negligible in 3D-1 and 3D-3 is that the nitro and amino forming hydrogen bond are not affected by the π-π interaction as they are not on the same directions.

Fig.8.The inter-IGM and electronic density difference diagrams of 3D-1~3D-3.
Corresponding to 3D-1,3D-2 and 3D-3,extra layer is introduced in 3D-4,3D-5 and 3D-6 so as to analyze the relationship among layers(Fig.7).For comparison,the systems are divided in two ways and the δEs have also been calculated and listed in Table 3 and Fig.S6.As same as the division for 3D-1,3D-2 and 3D-3,in first way the two molecules in middle layer are fragment I and II,and the two layers left is fragment III.In this moment,the δEs all become positive and the absolute value are enlarged on the basis of 3D-1,3D-2 and 3D-3,which manifests the extra layer brings extra anticooperative effect on the interaction between fragment I and II.According to the inter-IGM diagram,the interaction between layers is similar to that in 3D-1,3D-2,and 3D-3.Because of the simultaneous effect from both the top and bottom layers,a redistribution of electron on fragment I and II can also be observed,which leads to weakness hydrogen bonding in inner layer.
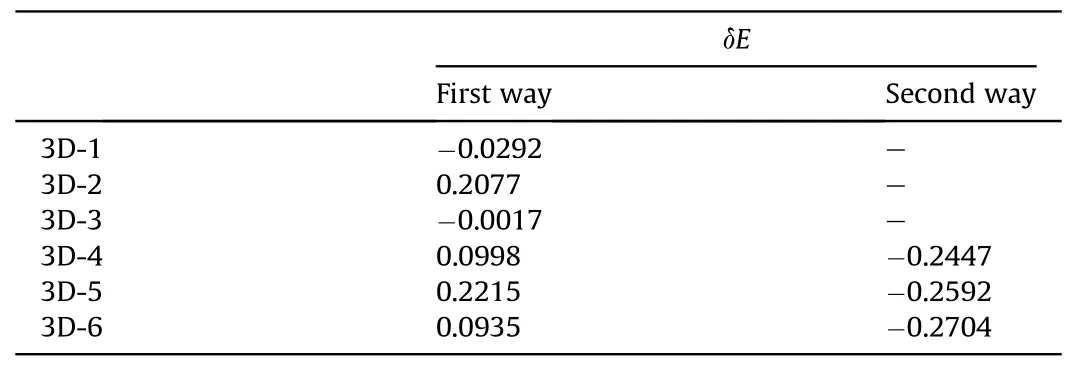
Table 3 The cooperative energy δE (kcal/mol) of 3D-1~3D-6 in two division ways.
In the second way,the three layers are divided into three fragments,respectively.Interestingly,the δEs change into negative,demonstrating a strong cooperativity relationship exists among layers.The reason is that the middle layer is treated as bridge to connect with the top and bottom one via the π-π interactions.Meanwhile,as the redistribution of electrons shows the same way with the interactions,they will accelerate each other to accomplish cooperativity.According to the comparison of the two division ways,the estimation of cooperativity is proved to be highly dependent on the way of analysis.There should be claimed that with the lack of an accurate method for global evaluation,it is still difficult to judge the cooperativity of a more complicated system.Considering the results above,the crystal stability can be understood as a term that balances cooperativity andanti-cooperativity effects.It is essentially in agreement with the conclusion obtained by Mahadevi,A.S.and coworkers[30].
4.Conclusions
In summary,we have proved the existence of cooperativity among the non-covalent interactions in TATB crystal.Its origin and mechanism have also been properly demonstrated.The cooperativity of typical non-covalent interactions in TATB crystals has been qualitatively and semi-quantitatively evaluated through the theoretical calculations,and the general rules have been revealed.With the assistance of cooperativity,the strength of non-covalent interactions shows apparent enhancement by promoting the interaction energy over 10%,which brings extra stabilization to improve the resistance of crystals to the external stimuli.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
The authors acknowledge the support from the National Natural Science Foundation of China (No.21875184),the Natural Science Foundation of Shaanxi Province (No.2022JC-10),and Youth Talent of Shaanxi“TeZhi” Program.
Appendix A.Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.dt.2022.04.019.

- Defence Technology的其它文章
- In-plane and out-of-plane quasi-static compression performance enhancement of 3D printed re-entrant diamond auxetic metamaterial with geometrical tuning and fiber reinforcement
- Flame behavior,shock wave,and instantaneous thermal field generated by unconfined vapor-liquid propylene oxide/air cloud detonation
- Frequency domain analysis of pre-stressed elastomeric vibration isolators
- Burning surface formation mechanism of laser-controlled 5-aminotetrazole propellant
- Blast disruption using 3D grids/perforated plates for vehicle protection
- Nonlinear tight formation control of multiple UAVs based on model predictive control