
异口恶唑环抗癌药物研究进展
2023-07-18 03:15:30孙文魏成喜卞明
系统医学 2023年6期
孙文,魏成喜,卞明
1.内蒙古民族大学医学院,内蒙古通辽 028000;2.内蒙古民族大学药物化学与药理学研究所,内蒙古通辽 028000;3.通辽市中医医院西药房,内蒙古通辽 028000
异噁唑是一类应用广泛的杂环化合物,已被证明是药物合成中用途非常广泛的构件,其生物活性包括抗癌、免疫调节[1-2]、抗阿尔茨海默病[3-4]、抗帕金森[5]、降血糖[6]、镇痛、抗炎[7]、抗菌和抑制HIV等诸多药理活性。随着研究的发展,异噁唑环已被用于制备小分子药物或天然产物结构修饰。它可以由含有肟结构的官能团与富含电子的炔烃通过氧化成环得到。目前,异噁唑类衍生物的药物较多,因其便捷的合成方式和优秀的生物活性受到药物化学学者青睐,尤其在抗癌治疗方面表现出了良好的应用前景[8]。现如今,异噁唑衍生物结构已成为有机化学家与药物化学家等众多科研人士研究的热点之一。随着新化合物不断涌现,在研究其化学结构及生物活性之后,分析并总结其规律,这样可以根据不同的需要选择合适的先导化合物。为了能够开发出更高效的抗癌新药,研究异噁唑类化合物是非常必要的。
1 抗癌活性
Im D等[9]设计和合成了一系列具有喹唑啉核心的4-芳基氨基5-甲基异噁唑衍生物。其中化合物1a(见图1),且对FLT3-ITD和FLT3-TKD(D835Y)有活性,IC50值分别为301 nM和228 nM。化合物1a显示出优异的选择性特征,对其他激酶包括FMS、cKit具有20%或更低的活性。分子对接结果显示1a可以对接至FLT3激酶的ATP结合位点。因此,化合物4a显示出作为急性白血病治疗剂的潜力。
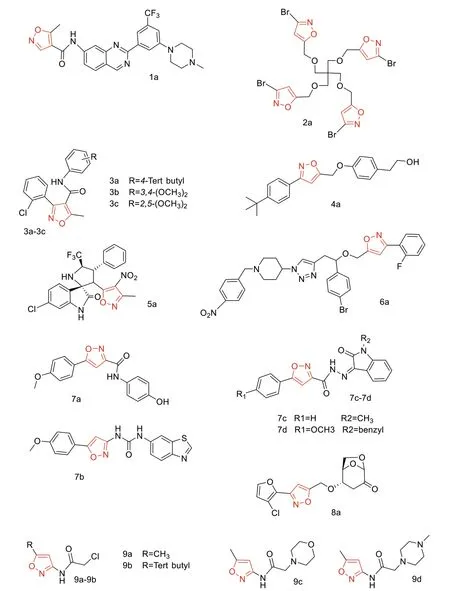
图1 药物化学结构式
Yazdani H等[10]用一种简单的合成方法,基于炔基和溴氰氧化物之间的1,3-偶极环加成反应,然后将亲水性聚乙二醇尾缝合到核上进行酯化反应,为合成含有异噁唑基枝状体的结构聚合物提供了1条新的途径。将聚乙二醇尾端加到端基树枝状分子中(见图1),使其在包括水介质在内的各种溶剂中具有溶解性,并使端基树枝状分子结构具有空间稳定性。脑肿瘤细胞(U251N)用浓度在100 µM时的化合物2a处理,在24、72 h后,化合物2a显著降低了活细胞数量,分别降至9.89%、1.35%。研究结果表明化合物2a通过抗肿瘤剂的共价和非共价连接,以及掺入细胞表面结合分子探针以提高细胞类型特异性,进一步探索它们联合治疗的潜力。
与常用的抗癌药物阿霉素(DOX)作比较,Eid AM等[11]合成了一系列异噁唑甲酰胺类化合物(见图1),并测定了它们对乳腺癌细胞(MCF-7)、宫颈癌细胞(HeLa)和肝癌细胞(Hep3B)的细胞毒活性和抗氧化活性。结果表明,化合物9b和9c对Hep3B细胞的抑制活性最强,IC50值约为23 µg/mL,9d对HeLa细胞的半数抑制浓度最高,IC50值为15.48 µg/mL。然而,化合物3a对MCF-7细胞具有最低的IC50值39.80 µg/mL)。化合物9b和9c均可使Hep3B分泌甲胎蛋白的量减少分别为(1 829.33±65.91)ng/mL和(1 758.66±54.04)ng/mL。此外,在细胞周期分析中,化合物3a和3b诱导的G2/M期延迟为18.07%,与阿霉素阳性对照相似。此外,化合物3b和3c可使Hep3B细胞的坏死率降低3倍,并使细胞发生凋亡。结果表明,化合物3b和3c具有较强的抗癌活性。
以天然酚类化合物酪醇为原料,Aissa I等[12]合成了3,5-二取代异噁唑衍生物(见图1)。酪醇是一种天然酚类化合物,广泛存在于橄榄油等天然来源中,具有广泛的生物活性。大多数衍生物对人胶质母细胞瘤癌细胞(U87)具有显著的抗增殖活性,且呈剂量依赖关系。化合物4a的IC50(15.2±1.0)µg/mL具有较强的抗癌活性。结果认为新合成的异噁唑连接的酪醇衍生物是开发有效抗癌药物的有前途的支架。
Liu S等[13]合成了一系列含CF3的高活性3'-(硝基异恶唑)螺(吡咯烷-3,20-氧吲哚)。将具有生物活性的螺辛吲哚和异噁唑骨架结合,得到的化合物5a(见图1),对这两个靶标均表现出很强的活性(Ki=112.5)。化合物5a在MCF-7乳腺癌细胞中表现出了良好的抑制MDM2介导的p53降解活性和GPX4水平的活性。此外,在体内实验中,化合物5a对MCF-7异种移植模型中MDM2和GPX4均表现出抑制作用,可触发嗜铁性和凋亡细胞死亡这与体外实验结果一致。
Murthy A等[14]设计并合成了一个含3-芳基异噁唑侧链的新型哌啶-三唑杂化化合物,见图1。细胞毒性研究发现,其中化合物6a是最具潜力的衍生物,对具有代表性的7种癌细胞株具有很高的活性。通过BRET分析,确定活癌细胞中PUMA/BclxL相互作用的IC50值为3.8 µM。通过分子模拟研究,带有芳基异噁唑部分的新侧链应该部分填充PUMA结合的BCL-XL凹槽。因此,相应的数据使该实验能够提出这个新分子的活性的理论基础,并确认这一家族化合物对PUMA-BCL-XL强相互作用的扰动潜力。
Abou-Seri SM等[15]合成了3个新系列的基于异噁唑的甲酰胺系列、尿酸盐系列和腙系列(见图1),并对其潜在的生长抑制活性进行了生物学评估60种癌细胞系。化合物7a对48种癌细胞系显示出广谱抗癌活性,平均GI=30.9%。抗肝细胞癌(HepG2)化合物的综合抗癌评估表明,尿酸盐衍生物7b和腙衍生物7c和7d通过促进细胞周期停滞和诱导细胞凋亡显示出比索拉非尼IC50值为3.99 µM更好的抗癌活性IC50值为0.84、0.79、0.69 µM。此外,还显示了化合物7b、7c和7d对THLE2正常肝细胞的有希望的安全性。血管内皮细胞生长因子受体2(vascular endothelial growth factor receptor-2, VEGFR-2)抑制活性的评估表明,化合物7b和16c是本研究中最有效的VEGFR2抑制剂,IC50值分别为25.7和28.27 nM。与VEGFR2结合位点的对接表明,所提出的衍生物产生了与索拉非尼相当的拟合值、结合能和结合相互作用。因此,化合物7b、7c和7d显示出高体外效力和对HepG2癌细胞的良好选择性、有效的VEGFR-2激酶抑制作用并预计具有良好的药代动力学特征,代表了探索其在癌症治疗中潜在益处的有吸引力的工具。可以得出结论,该研究强调了所提出的设计策略,并提出了一种新的分子支架,可以进一步优化并用于设计有前途的强效VEGFR-2抑制剂。
通过杂化Michael加成和1,3-偶极环加成序列,Tsai Y等[16]设计合成了新型左旋糖苷酮类衍生物(见图1)。所有异噁唑都具有细胞毒性,其IC50值范围为13.8~26.4 µM。这些值低于相对应1,2,3-三氮唑取代衍生物的结果,表明侧链上的异噁唑环为更突出的结构基序。这可以通过比较芳香族片段上具有相同取代基的化合物观察到的活性来证明,这些异噁唑的活性(平均)比相对应的三唑类似物高43%。合成的活性最高的化合物是8a,具有5-氯-2-呋喃取代基,其IC50值为13.8 µM。
Warda ET等[17]合成了一系列新的异噁唑类化合物(见图1、图2)并对其体外抗肿瘤活性进行了评价。结果表明,化合物9b和9i对3种癌细胞的抑制作用最强,IC50值为6.38、9.96 µM。此外,化合物9a、9d和9g对这3种癌细胞表现出较强的活性,而化合物9c、9e、19f和9g对这3种癌细胞表现出中等活性。此外,化合物9i对Wish和WI38正常细胞具有较低的细胞毒作用的IC50值分别为(53.19±3.1)、(38.64±2.8)µM,有望成为一种安全有效的抗肿瘤药物。研究了9个活性化合物9a~9i对EGFR-TK的抑制活性,其中化合物9e、9f和9i的抑制活性最高的IC50值分别为(0.064±0.001)、(0.066±0.001)和(0.054±0.001)µM。化合物9i还与其他4个靶蛋白进行了比较,结果表明,化合物9a对VEGFR-2、细胞角蛋白2α(CK2α)和拓扑异构酶Ⅱβ具有良好的抑制活性,对微管蛋白聚合具有较好的抑制活性。细胞周期分析表明,9i诱导癌细胞周期停滞于G2/M期和前G1期。此外,还证实了9i通过caspase3/9水平升高和Bax/Bcl2比值升高而诱导癌细胞死亡。对接研究证明,化合物9i与EGFR-TK、VEGFR-2、CK2α、拓扑异构酶IIβ和微管蛋白的活性部位完全匹配。利平斯基规则和Veber标准也进行了分析,结果表明化合物9i有望被口服吸收。

图2 药物化学结构式
用钯交叉偶联的方法Weaver MJ等[18]合成了一系列含N杂环(N-het)的蒽基异噁唑酰胺(AIM)类抗肿瘤药物10a、10b(见图2)。对脑癌细胞和乳腺癌细胞都表现出很强的抗肿瘤活性。在这两种情况下,化合物20b是最活跃的化合物。对于乳腺癌细胞株MDA-468,所有化合物都表现出个位数的微摩尔细胞毒性,其中化合物10a与10b大致相当。在脑瘤细胞系SNB-19中,化合物10b的活性明显高于其他杂环,化合物10a的活性为2 µM。此外,还检查了大鼠胶质瘤细胞系,N-杂环AIMS对其活性较低,然而,Quin-AIM确实在6.71 µm处保持了个位数的微摩尔效价,这表明该细胞系可能被用于AIMS的临床前表征的动物模型中。
Wu X等[19]所在课题组通过高通量虚拟筛选(HTVS)方法确定了一个潜在的乙酰辅酶A羧化酶(acetyl coA carboxylase, ACC)抑制剂11a,并合成了一系列4-苯氧基苯基异噁唑类化合物(见图2)用于SAR研究。其中,化合物11b的抑癌活性最强的IC50值为99.8 nM,与CP-640186相当。抗增殖实验表明,化合物11c的细胞毒性最强,其IC50值分别为0.22 mM(A549)、0.26 mM(HepG2)和0.21 mM(MDA-MB-231)。化合物11b和11c的初步机制研究表明,这些化合物降低了丙二酰辅酶A水平,使细胞周期停滞在G0/G1期,并诱导了MDA-MB-231细胞的凋亡。这些结果表明4-苯氧基苯基异噁唑作为ACC抑制剂在癌症治疗中具有进一步研究的潜力。
Zhu P等[20]设计合成了一系列新型的含异噁唑的联苯类化合物(见图2),并对其作为PD-1/PD-L1抑制剂进行了评价。构效关系表明,引入异噁唑的闭环策略是可行的,并且3-氰基苯基对PD-1/PD-L1蛋白-蛋白质相互作用具有显著的抑制活性。分子对接研究有助于了解小分子抑制剂与PD-L1二聚体的结合模式。特别是化合物12a是1种很有前途的抗PD-1/PD-L1抑制剂,其IC50值为23.0 nM,为今后的药物开发提供了有价值的信息。
2 结论
本文综述了近几年异噁唑衍生物的抗癌研究进展并归纳了它们的生物活性、构效关系、治疗作用和化学结构。对不同类型化合物进行分析比较后发现它们都具有较好的抗肿瘤活性;在体外实验结果表明它们均能有效抑制癌细胞生长;在体内外动物试验表明它们对肿瘤细胞也表现出良好疗效。但存在一些问题需要解决。作者认为异噁唑结构中N、O原子能够与受体形成增强结合力的H键,异噁唑本身具有芳香性能够形成π-π共轭,这使异噁唑成为重要的生物活性官能团。综上所述,异噁唑是非常具有抗癌潜力的结构片段,值得今后进一步研究。
猜你喜欢
Journal of Traditional Chinese Medicine(2022年3期)2022-07-20 15:53:38
抗癌之窗(2020年1期)2020-05-21 10:18:10
特别健康(2018年9期)2018-09-26 05:45:26
中学生数理化·高二版(2016年3期)2016-12-26 09:36:58
国外医药(抗生素分册)(2016年3期)2016-07-12 14:25:08
合成化学(2015年10期)2016-01-17 08:56:26
华东师范大学学报(自然科学版)(2014年1期)2014-04-16 02:54:58
无机化学学报(2014年10期)2014-02-28 17:33:13
中学生数理化·八年级数学华师大版(2008年3期)2008-08-26 11:26:16
现代农业科技(2006年23期)2007-01-05 03:23:54
