
基于生物信息学方法筛选系统性红斑狼疮疾病进展中与先天免疫细胞相关的关键基因
2023-06-30 09:26左旭李津
天津医科大学学报 2023年3期
左旭,李津
(天津医科大学基础医学院生物信息学系,天津 300070)
系统性红斑狼疮(systemic lupus erythematosus,SLE)是一种最为常见的自身免疫性疾病。成年女性的发病率约为男性的8 倍,其发病累及全身多种器官,对患者的身心健康造成了巨大的危害[1]。SLE 的发病和进展是遗传和环境因素共同作用的结果,并且受到雌激素水平的影响。有研究报道SLE 患者的一级亲属患病风险比健康人群高20 倍[2];环境因素,如紫外线和阳光照射会导致细胞凋亡增加,进而释放富含自身抗原的细胞外囊泡,从而对SLE 的发病有一定的影响[3];性别和激素影响也是SLE 的危险因素之一,已有研究发现,雌激素会通过刺激B 细胞增殖和随后的自身抗体产生来促进体液免疫反应。
目前,对SLE 发病机制的主要研究集中于致病性自身抗体及免疫复合物、T 细胞和自然杀伤(NK)细胞功能失调等方面[4]。总的来说,SLE 的发展主要是在各种病因的影响下导致致病性自身抗体的产生;另一方面部分SLE 患者由于CD8+T 细胞和NK细胞的功能失调,不能抑制CD4+T 细胞的持续作用,导致B 细胞持续活化产生自身抗体,继而与细胞组织或循环中的抗原结合,形成免疫复合物通过沉积在各器官的组织中或直接引起组织和器官的损伤。先天性和适应性免疫系统在SLE 的发病机制中都发挥作用[5-6]。异常的先天免疫反应在SLE 的发病机制中起重要作用,其通过释放炎性细胞因子导致组织损伤以及自身反应性T 和B 细胞的异常激活,进而导致致病性自身抗体的产生和终末器官伤害。而SLE 患者中先天免疫细胞在狼疮发病进展中的作用和机制还缺乏深入研究[7-8]。探索SLE 发生、发展的分子特征和机制,鉴定出与SLE 相关的遗传学特征,为SLE 的有效预防、诊断和治疗提供新的策略显得非常重要。
微阵列数据(microarray data)以及RNA 高通量测序(RNA-sequencing,RNA-seq)数据已广泛用于在基因组水平上探索和鉴定用于疾病早期诊断和预后的生物标志物[9]。有遗传学研究表明,先天免疫过程在SLE 发病机制中也发挥重要作用[10]。例如,中性粒细胞溶解因子1(NCF1)和NCF2 基因中的错义单核苷酸多态性(SNPs)与SLE 疾病相关[11];单核细胞上FcγRI/CD64 的表达增加与SLE 的持续炎症和肾炎相关[12]。但对于SLE 的先天免疫过程中的关键基因了解还不够充分。因此在本研究中,基于微阵列数据以及RNA-Seq 数据,在转录水平分析出先天免疫细胞的异常与SLE 的发病高度相关,并通过加权共表达网络分析(Weighted Gene Co-ex pression Network Analysis,WGCNA)构建了共表达网络,结合免疫细胞组成和丰度分析,筛选出与SLE先天免疫相关的枢纽基因,为SLE 的早期诊断及治疗提供一定的帮助。
1 材料与方法
1.1 数据来源及预处理 从高通量基因表达数据库(Gene Expression Omnibus data base,GEO)中共收集来自GSE50772、GSE81622 和GSE99967 的89例SLE 患者血液样本和62 名健康对照血液样本的基因表达微阵列数据,作为本研究的发现队列。分别使用R(版本3.5.2)中Affy 包的rma(Robust Multiarray Average)行归一化,并且对于有多个探针或探针组的基因取中位数的值。然后,采用KNearest Neighbors 的方法来处理3 个数据集中的缺失值。之后,首先使用R 软件包的inSilicoMerging 来合并数据集,然后,使用sva 包的combat 功能去除批次效应。选取GPL169791 Illumina HiSeq 2500(Homo sapiens)和GPL18573 Illumina NextSeq 500(Homo sapiens)平台的GSE122459 数据集作为验证队列,其包括20 例SLE 患者血液样本和6 名健康对照血液样本。
1.2 免疫细胞组成和丰度分析 采用CIBERSORT和xCELL 方法对这89 例SLE 患者数据进行分析,CIBERSORT 采用反卷积算法通过具有22 种免疫细胞亚型的参考集(LM22)估计免疫细胞组成和丰度,xCELL 采用ssGSEA 算法通过计算样本在每种细胞类型标记的富集分数并将其转换为细胞类型分数,最后进行校正得到各细胞类型的xCELL 评分[13]。之后,筛选出先天免疫细胞并采用秩和检验来比较SLE 组和对照组之间的免疫细胞组成差异,使用R 中的ggplot2 包来进行可视化。
1.3 加权基因共表达网络的构建 通过利用89 例SLE 样本的基因表达谱,剔除方差最小的前70%的基因,得到3 689 个基因,然后,使用R 中的WGCNA 包来构建共表达网络。首先,基于配对基因之间的Pearson 相关值,将单个转录本的表达水平转换为相似矩阵。接下来,将相似度矩阵转换为邻接矩阵,计算公式为amn=|cmn|β(cmn=配对基因之间的Pearson 相关性;amn=配对基因之间的邻接)。参数β可以提高基因间的强相关性,降低基因间的弱相关性。选择β=9 的幂时,邻接矩阵转换为拓扑重叠矩阵(Topological overlap matrix,TOM)。用基于TOM不同性测量的平均连杆分层聚类对显示相似表达谱的基因进行分类,这些基因模块由簇树的分支和不同颜色表示[14]。计算模块特征基因与先天免疫细胞组成和丰度水平之间的相关性,通过Pearson 检验确定模块的显著性。
1.4 富集分析 使用R 软件的clusterprofile 包进行GO(Gene Ontology)和KEGG(Kyoto Encyclopedia of Genes and Genomes,京都基因与基因组百科全书)途径富集分析,以检测与关键模块基因相关的生物功能和潜在信号通路途径。GO 分析确定生物过程(biological process,BP)、细胞组分(Cellular component,CC)、分子功能(molecular function,MF),P<0.05 为筛选标准。KEGG 分析确定潜在信号通路途径,FDR<0.05 作为统计显著性标准。
1.5 差异表达基因分析及关键基因的筛选 健康样本和SLE 样本间的差异表达基因(differentially expressed gene,DEG)是通过R 软件包limma 来识别的,以|log2 FoldChange|≥1 和FDR<0.05 作为有统计学意义的差异标准进行筛选。计算与基因的表达相关性以获得基因显著性(gene significance,GS),同时计算模块特征向量与基因的表达相关性以获得模块成员关系(module membership,MM),选取了MM>0.8,且GS<0.6 的基因作为枢纽基因。此外,在关键模块和DEG 中的基因之间重叠了基因以进一步确定关键基因。
1.6 基因集富集分析(gene set enrichment analysis,GSEA) 根据关键基因的表达水平的中位数将样本分成高表达组和低表达组,以MSigDB 数据库的c2.cp.kegg.v7.4.symbols.gmt 为参考基因集,用以评估相关途径和分子机制,基于基因表达谱分组,设定最小基因集为5,最大基因集为5 000,经1 000次重抽样,以|NES|>1,P<0.05,FDR<0.25 为明显富集基因集的标准。
1.7 受试者工作特征(ROC)分析 利用R 软件的pROC 包进行SLE 诊断中具有高灵敏度和特异性的关键基因的鉴定。并在GSE122459 数据集中计算了关键基因的表达与疾病中性粒细胞比例的斯皮尔曼相关系数。
2 结果
2.1 去批次效应以及免疫细胞组成和丰度分析 将数据集进行整合,并进行去除批次效应(图1),然后通过CIBERSORT 和xCELL 进行免疫细胞组成和丰度分析,将先天免疫细胞的结果进行可视化。CIBERSORT 的分析发现SLE 组与对照组相比,静息的NK 细胞和静息的肥大细胞的比例减少,而单核细胞、巨噬细胞M0、活化的树突状细胞和中性粒细胞的比例增加(图2A);xCELL 的分析显示,SLE组中同样是NK 细胞和肥大细胞的比例较对照组少,而单核细胞、巨噬细胞、树突状细胞和中性粒细胞的比例更多(图2B)。

图1 去批次效应UMAP 图Fig 1 UMAP diagram before and after removing batch effect
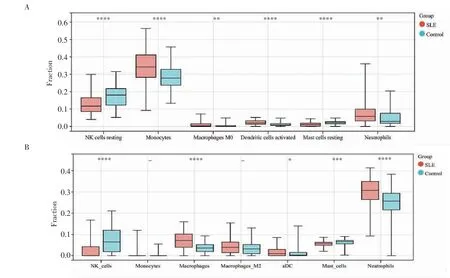
图2 免疫细胞组成和丰度分析Fig 2 Immune cell composition and abundance analysis
2.2 WGCNA 构建共表达网络 选取拟合指数R2为0.86 时的软阈值β=9(图3A),基于选择的软阈值构建共表达网络,共识别到13 个模块,并通过层次聚类树来展示所构建的13 个模块(图3B)。

图3 软阈值的确定及共表达网络构建Fig 3 Determination of soft thresholds and co-expression network construction
2.3 免疫细胞相关模块的确定及其KEGG 通路分析 将免疫细胞组成和丰度分析中均具有统计学意义的结果作为性状与模块内的基因相关联,结果显示棕褐色模块与静息的NK 细胞(cor=0.80,P<0.05)和绿色模块与中性粒细胞(cor=0.85,P<0.05)具有显著相关性(图4),故将绿色模块和棕褐色模块作为关键模块进行后续分析。

图4 模块基因与性状相关性图Fig 4 Correlation between module genes and traits
2.4 富集分析 对棕褐色模块的基因和绿色模块的基因进行KEGG 富集分析,绿色模块基因富集在白细胞介素(IL)-17 信号通路、核因子(NF)-κ B 信号转导途径、肿瘤坏死因子(TNF)信号转导通路等(图5A),棕褐色模块基因富集在抗原处理和呈现、NK 细胞的细胞毒性等免疫系统相关的通路上(图5C)。对棕褐色模块和绿色模块的基因进行GO 通路富集分析,绿色模块基因主要参与免疫反应、趋化作用和炎症等生物途径;在细胞组分(CC)中主要富集到分泌颗粒膜、分泌囊泡等;在分子功能(MF)中主要富集在细胞因子活性、信号受体结合等活动上(图5B)。棕褐色模块基因在生物过程(BP)中主要富集到免疫反应、细胞杀伤等;CC 分析表明,这些基因产物的位置主要富集在细胞溶解颗粒;MF 富集显示,它们的分子功能(MF)主要富集到MHC Ⅰ类受体活性等(图5D)。

图5 KEGG 和GO 富集分析Fig 5 KEGG and GO enrichment analysis
2.5 枢纽基因的筛选 计算绿色模块和棕褐色模块内基因的GS 和MM 值并绘制散点图,筛选了MM>0.8、GS>0.6 的基因作为枢纽基因,其中绿色模块中有15 个枢纽基因(图6A),棕褐色模块中有8个枢纽基因(表1 和图6)。

表1 绿色模块和棕褐色模块中的枢纽基因Tab 1 Hub genes in green module and tan module

图6 关键模块基因相关性散点图Fig 6 Module eigengenes in the key module
2.6 差异表达基因分析及关键基因的筛选 对合并的数据集进行差异表达基因分析,得到上调差异基因45 个,下调差异基因5 个(图7A)。将这些差异表达基因与筛选得到的枢纽基因整合,在绿色模块中得到两个关键基因,分别是CXCL1(C-X-C Motif Chemokine Ligand 1)和MME(Membrane Metalloendopeptidase)(图7B)。
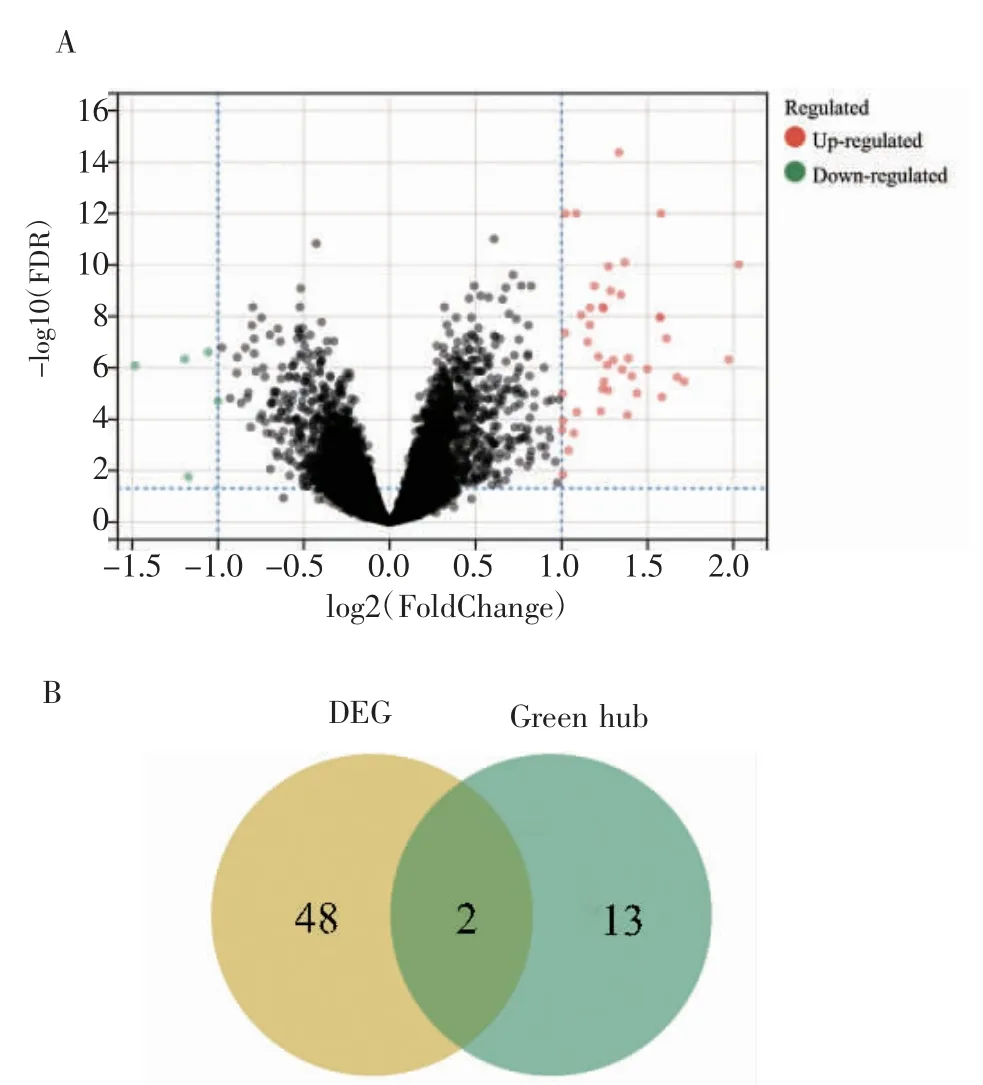
图7 差异基因表达与关键基因筛选Fig 7 Differential gene expression and key gene screening
2.7 GSEA 分析 结果表明,CXCL1 富集到NOD样受体信号通路(NES=1.97,P =0.002)、利什曼病的感染通路(NES=1.87,P=0.004)且均与其表达正相关,而硒胺酸的代谢通路(NES=-1.83,P=0.006)与其负相关(图8A)。MME 富集到叶酸的代谢(NES=1.79,P=0.002)、Toll 样受体信号通路(NES=1.82,P=0.002)和NOD 样受体通路(NES=1.82,P=0.002)且均与MME 的表达呈正相关(图8B)。

图8 基因集富集分析Fig 8 Gene set enrichment analysis
2.8 GSE122459 数据集验证 使用GSE122459 数据集对两个关键基因CXCL1 与MME 进行验证,在表达上,两个基因在SLE 患者中表达量升高(图9A),CXCL1 基因的AUC(Area under curve)值为0.86,MME 基因的AUC 值为0.81(图9B)。对数据集进行免疫细胞比例分析后,计算其表达与中性粒细胞相关性,CXCL1 的表达与中性粒细胞的相关性为0.79(图9C),MME 的表达与中性粒细胞的相关性为0.87(图9D)。
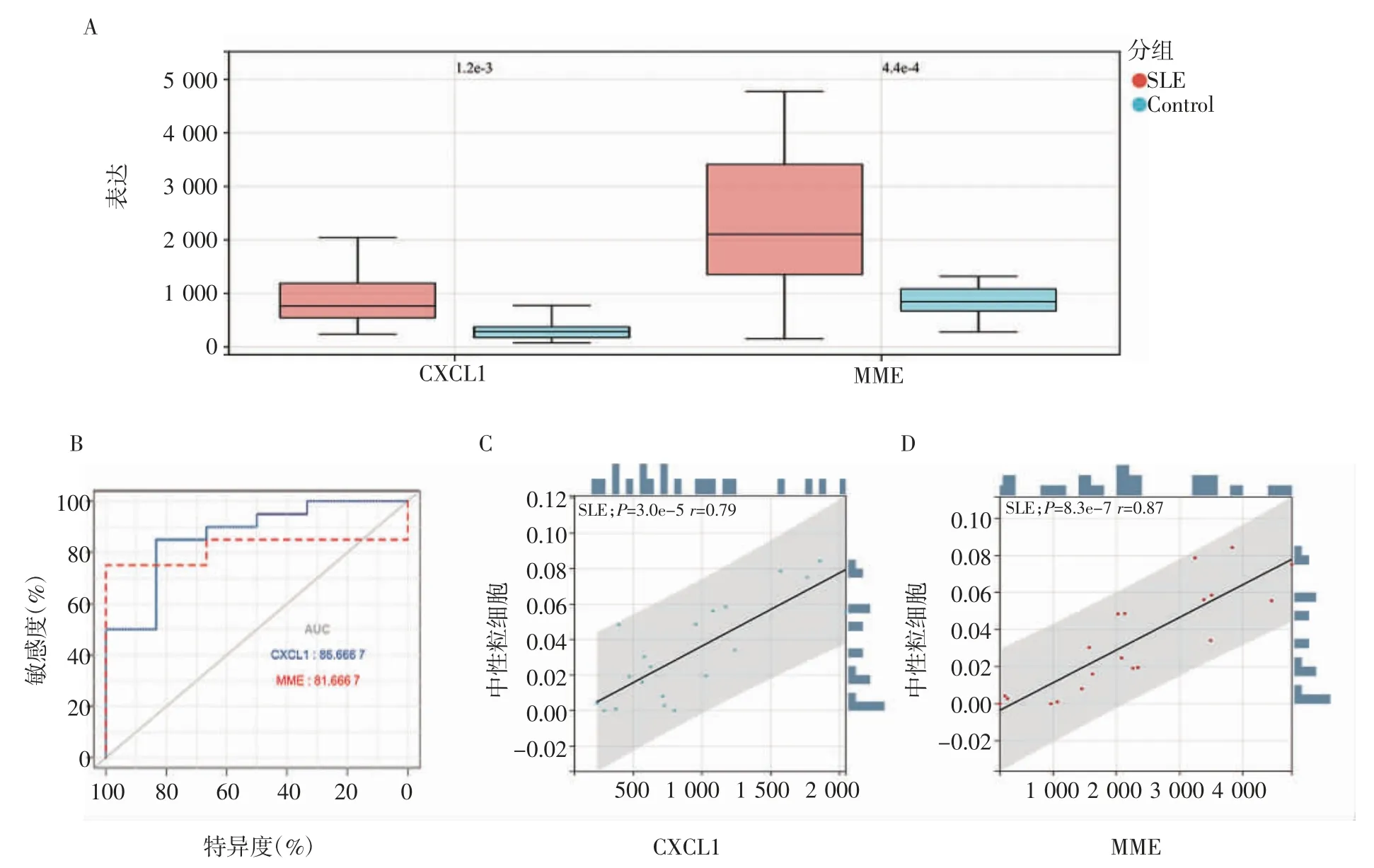
图9 CXCL1 和MME 在GSE122459 数据集中的验证Fig 9 Validation of CXCL1 and MME in GSE122459 dataset
3 讨论
SLE 具有高度复杂性和异质性。患者免疫系统多组成部分出现明显的功能障碍,并且呈现临床异质性,所以对SLE 的精准治疗面临着巨大的挑战。关于SLE 发病过程中先天免疫系统的作用机制的报道较少,但最近有研究发现中性粒细胞铁死亡对于人自身免疫性疾病有着重要作用,证实了先天免疫细胞异常会导致系统性自身免疫性疾病[15]。因此,开展对于SLE 发病过程中先天免疫的作用机制的研究具有重要意义。
本研究整合了GSE50772、GSE81622 和GSE99967数据集,进行了先天免疫细胞组成和丰度的全面评估,发现NK 细胞和肥大细胞在SLE 中的比例减少,而单核细胞、巨噬细胞、树突状细胞和中性粒细胞在SLE 中的比例较对照组增加,提示这些细胞类型在SLE 的发生和发展中发挥重要作用。之前的研究表明,NK 细胞的细胞毒性功能在SLE 患者中受损[16];肥大细胞及其激活相关抗体参与了类风湿性关节炎和多发性硬化症等各种自身免疫性疾病的发生和发展[17];低密度粒细胞(LDG,low-density granulocytes)即中性粒细胞的一种促炎性中性粒细胞亚群与狼疮的特定临床特征的存在和严重程度相关等[18]。
为了进一步鉴定与疾病相关的模板,对数据集进行了WGCNA 分析,共得到13 个基因共表达模块,并鉴定了1 个与NK 细胞高度相关的模块即棕褐色模块和1 个与中性粒细胞高度相关的模块即绿色模块。与NK 细胞相关的模块基因主要富集中在细胞溶解颗粒、细胞杀伤、抗原处理和呈现、NK细胞的细胞毒性等通路中;与中性粒细胞相关的模块基因主要富集于细胞因子活性、IL-17 信号通路、NF-kappa B 信号传导途径等。有研究证实,对细胞因子的监测能够提高确定SLE 疾病活性的敏感性和特异性,如通过IL-18 能够预测活动性肾脏SLE的风险,而IL-6 和IL-8 能够预测活动性非肾脏的风险[19]。中性粒细胞在SLE 中发挥至关重要的作用。狼疮相关免疫复合物在FcγRIIA 依赖但非TLR的响应中激活人类中性粒细胞[20],中性粒细胞外陷阱NET 中的mtDNA、抗mtDNA 抗体与SLE 中的PDC IFNα 发病机制之间相关联[21]。
之后,结合DEG 分析选择CXCL1 和MME 作为与中性粒细胞相关的关键基因。GSEA 分析发现,CXCL1 和MME 都有富集到NOD 样受体信号通路。近期有研究报道NOD 样受体信号通路及其通路相关基因在SLE 中的重要作用,NOD 样受体含吡啶域蛋白3(NLRP3)炎症体的失调在系统性红斑狼疮中起着重要作用,高水平的let-7f-5p 可以通过靶向NLRP3 来减轻SLE 炎症[22]。此外,CXCL1 的表达还可能与利什曼病的感染通路正相关并与硒氨酸的代谢呈负相关,内脏利什曼病(Visceral leishmaniasis,VL)与SLE 的症状有很强的相似性[23];体内的许多硒蛋白参与内源性抗氧化防御系统,硒蛋白的水平低与各种疾病的发展密切相关[24]。MME 的表达还与叶酸的代谢和Toll 样受体通路呈正相关。有研究表明,低水平的叶酸与高浓度的同型半胱氨酸相关,而高同型半胱氨酸血症是SLE 患者亚临床动脉粥样硬化的独立风险因素之一[25-26]。网状或凋亡中性粒细胞释放的核酸通过病毒核酸特异性Toll 样受体激活先天和适应性免疫是狼疮肾炎的发病机制之一[27]。
然后,基于GSE122459 数据集进行了ROC 分析以及基因的表达与中性粒细胞的相关性分析来评估基因与患者诊断之间的相关性。CXCL1(AUC=0.86)和MME(AUC=0.81)均在SLE 诊断中表现出较高的灵敏度和特异性,且其表达均与中性粒细胞有较高的相关性。CXCL1 属于CXC 趋化因子家族的一员,可作为多种免疫细胞的趋化剂,是一种有效的中性粒细胞趋化剂和激活剂[28],同时,CXCL1也能作为将活跃的白细胞招募到炎症组织的重要原因之一。已有研究报道,CXCL1 表达水平的变化可能是SLE 活动的潜在标志[29]。MME 编码膜金属内肽酶也称中性内肽酶,主要存在于肾脏、肺、大脑和肝脏细胞的血浆膜中。尽管目前没有其在SLE 中的研究,但有研究表明在与SLE 相关的膜性肾病中,中性内肽酶是重要的自身抗原之一,在调节炎症方面至关重要[30]。因此,推测CXCL1 和MME 可能在SLE 的疾病进展中发挥重要作用。本研究存在一定的局限性,由于使用公共数据进行分析,对于SLE的临床异质性无法获取全面的组织数据进行分析,从而选择了外周血的数据进行分析。外周血是SLE免疫系统的主要途径,外周血单核细胞(PBMC)是最先启动对目标器官的自身免疫过程的免疫细胞,因此,PBMC 的基因表达特征能够在一定程度上揭示目标器官中免疫细胞的分子特征。在本研究所揭示的关键基因还需要在更全面的组织样本队列中进一步验证[31]。
综上所述,本研究基于免疫细胞组成与WGCNA 识别了2 个与SLE 先天免疫相关的关键模块,以及2 个与中性粒细胞相关的关键基因即CXCL1和MME,在SLE 发生、发展的先天免疫中可能发挥着重要作用,值得深入研究。
猜你喜欢
中老年保健(2022年1期)2022-08-17
中国民间疗法(2021年14期)2021-08-30
中学生数理化(高中版.高考理化)(2021年6期)2021-07-28
英语学习(上半月)(2019年9期)2019-10-10
米娜·女性大世界(2016年8期)2016-08-17
工业设计(2016年11期)2016-04-16
医学研究杂志(2015年3期)2015-06-10
中国当代医药(2015年21期)2015-03-01
食品工业科技(2014年11期)2014-03-11
中国商人(2013年1期)2013-12-04
