
脂肪因子在糖尿病认知功能障碍中的调控作用及运动干预机制*
2023-06-20 04:42宗博艺李世昌
生物化学与生物物理进展 2023年6期
宗博艺 李 琳 李世昌 孙 朋
(1)华东师范大学青少年健康评价与运动干预教育部重点实验室,上海 200241;2)华东师范大学体育与健康学院,上海 200241)
《美国医学会杂志》(JAMA)发表的最新研究数据显示,在2013~2018年,中国成年人糖尿病的患病率已从10.9%增至12.4%。其中,70岁以上老年人糖尿病的患病率已从20.7%增至27.3%[1]。除增加患心血管疾病、肾病和癌症的风险外[2],老年人罹患糖尿病尤其是2型糖尿病(type 2 diabetes mellitus,T2DM)还可诱发认知功能障碍[3-4],与阿尔茨海默病(Alzheimer's disease,AD)和血管性痴呆(vascular dementia,VD)等疾病的发生风险增加有关[5-6]。T2DM 诱发认知功能障碍的潜在机制包括高血糖、胰岛素抵抗、慢性低度炎症和氧化应激等[3,7]。随着中国人口老龄化进程加快,老年人患T2DM合并认知功能障碍将给社会和家庭带来沉重负担。因此,迫切需要推进糖尿病认知功能障碍诊疗策略的制定与实施。
脂肪因子(adipokine)是主要由白色脂肪组织(white adipose tissue,WAT)合成并分泌的一类细胞因子,可随血液循环进入大脑、骨骼肌和肝脏等组织器官,发挥调节食欲、胰岛素敏感性和炎症等多种生物学功能[8]。有研究发现,脂肪因子尤其是脂联素(adiponectin,APN) 和瘦素(leptin,LEP)还可参与调控个体认知功能,并可能是糖尿病认知功能障碍的生物标志物[9]。运动作为一种见效快、操作简便且无副作用的非药物干预手段,能够通过调节脂肪因子的生理水平改善肥胖和糖尿病等代谢综合征的能量代谢紊乱[10]。值得注意的是,适度运动也能够缓解糖尿病诱发的认知功能障碍,但具体机制尚不明确。本研究以“脂-脑”轴理论为切入点,深入探讨运动通过调节脂肪因子改善糖尿病认知功能障碍的可能性,为后续治疗靶点的筛选以及运动疗法的施用提供理论依据。
1 脂肪因子与糖尿病认知功能障碍的关系
1.1 脂联素与糖尿病认知功能障碍
APN 是人体血浆中含量最丰富的脂肪因子,属于补体1q 家族,包含244 个氨基酸,主要由WAT 合成并分泌,在循环中以高、中和低分子质量异构体的形式存在[11]。目前,已发现的脂联素受 体 (adiponectin receptor, AdipoR) 包 括AdipoR1、AdipoR2 和T-cadherin。其中,AdipoR1和AdipoR2 广泛分布于中枢神经系统内的下丘脑、皮层和海马等部位,在神经干细胞(neural stem cell,NSC)、神经元、小胶质细胞和星形胶质细胞中均有表达[12]。
人体研究发现,循环APN 的水平与老年人的整体认知功能、海马体积和脑白质病变的严重程度呈显著相关[13-15],并可作为老年T2DM患者认知功能障碍的早期诊断和治疗特异性和敏感性的生物标志物[16]。与对照组相比,T2DM 患者体内循环APN 的水平显著降低,而且与海马体积、灰质体积和脑葡萄糖代谢等指标[17-18]以及轻度认知障碍(mild cognitive impairment,MCI)的发生发展紧密 相 关[9,19-20]。动 物 研 究 发 现,APN基 因 敲 除(APN-/-)的老年小鼠出现脑胰岛素抵抗、认知缺陷和AD 样病理[21-22]。而APN 干预(0.1 µg/g)可通过激活磷脂酰肌醇3 激酶(phosphatidylinositol 3 kinase, PI3K)/磷 酸 激 酶B (phosphorylase kinase B,PKB,也称AKT)/糖原合成酶激酶3β(glycogen synthase kinase 3β,GSK-3β)信号通路以发挥神经保护作用,减弱tau 蛋白过度磷酸化,改善糖尿病大鼠的认知缺陷[23]。综上,APN 可能作为认知功能的重要调控器,缓解糖尿病相关的认知功能障碍。
1.2 瘦素与糖尿病认知功能障碍
LEP是由ob基因编码的一种含167个氨基酸的分泌型蛋白质,分子质量为14~16 ku,主要由WAT 合成并分泌。目前,已发现的瘦素受体(leptin receptor,LepR)有6 种,分别为LepRa、LepRb、LepRc、LepRd、LepRe 和LepRf。它们在中枢神经系统内多分布于下丘脑、海马、杏仁核和小脑等部位[24]。LEP与LepR结合后,磷酸化激活Janus 蛋白酪氨酸激酶2(Janus kinase 2,JAK2),进而活化下游的信号转导和转录激活因子3(signal transducer and activator of transcription 3,STAT3)、 PI3K 和细胞外信号调节激酶(extracellular regulated protein kinase, ERK) 等信号[25]。
在正常生理状态下,LEP是海马中重要的认知增强剂[26]。动物研究发现,LEP和LepR基因缺失(如ob/ob、db/db和fa/fa)的啮齿类动物表现出明显的海马依赖性认知缺陷[27]。然而,在T2DM 的病理状态下,LEP 水平异常升高导致其敏感性降低,造成中枢和外周瘦素抵抗,促进糖尿病认知功能障碍发展。与对照者相比,老年男性T2DM患者循环LEP的水平显著升高,且与认知功能呈显著负相关[28],循环LEP 水平异常升高以及LEP/APN 的比值增加是T2DM 患者合并MCI 的危险因素[29]。此外,也有研究发现,老年T2DM 合并MCI 患者循环LEP的水平与白介素-1β(interleukin 1β,IL-1β)的水平有关[9],这提示过高水平的LEP 可能参与介导T2DM相关的炎症。综合上述证据表明,尽管LEP也能够调控认知功能,但在不同生理状态下的情况有所不同。LEP的水平过低可能难以发挥神经保护作用,而水平过高则可导致瘦素抵抗和胰岛素抵抗,增加患认知功能障碍的风险。
2 脂肪因子调控认知功能的生物学机制
糖尿病导致的大脑微环境变化可能是认知功能障碍的重要诱因。APN 和LEP 不仅能调节血糖和胰岛素敏感性,也可穿过血脑屏障(blood-brain barrier,BBB)进入大脑[12,30],直接结合神经元和神经胶质细胞(如小胶质细胞和星形胶质细胞)膜上的受体,激活或抑制胞内下游信号通路,调节海马神经发生、突触可塑性、神经炎症、氧化应激和神经元凋亡等进程, 进而调控认知功能(图1)。
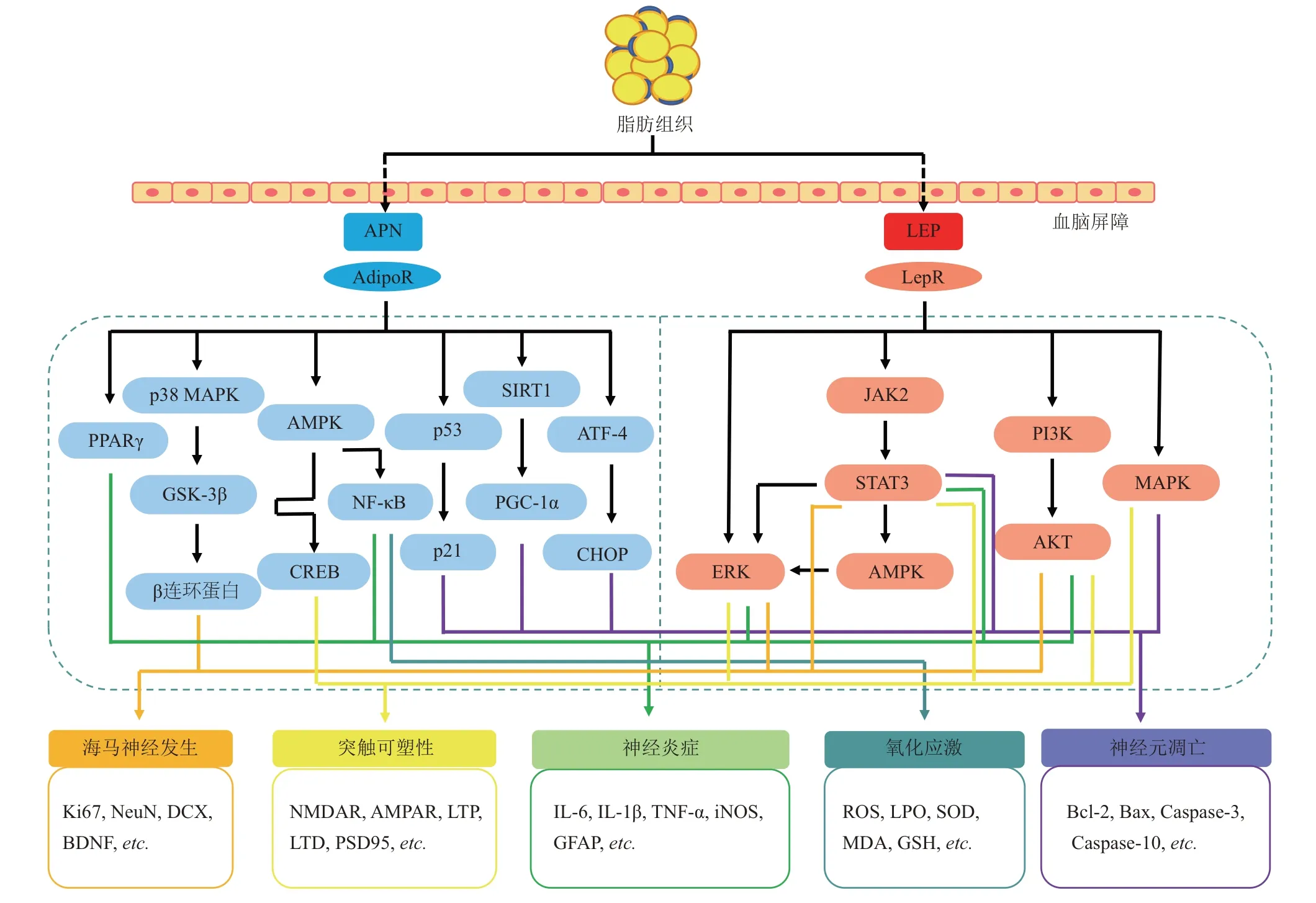
Fig. 1 The biological mechanism of adiponectin and leptin in regulating cognitive function图1 脂联素和瘦素调控认知功能的生物学机制
2.1 海马神经发生
海马神经发生(hippocampal neurogenesis)在学习和记忆过程中扮演重要角色。研究认为,糖尿病认知功能障碍可能是由于海马神经发生受损,海马新生神经元的数量减少导致的[31]。与对照组相比,高脂饮食(high-fat diet,HFD)结合链脲佐菌素(streptozotocin,STZ)诱导的T2DM 小鼠皮层和海马内Ki67、双皮质素(doublecortin,DCX)和神经元特异性核蛋白(neuron-specific nuclear protein,NeuN)的表达量减少,行为学表现出空间学习和记忆能力显著下降[32]。研究表明,与野生型小鼠相比,APN-/-小鼠海马齿状回(dentate gyrus,DG)内NSC增殖和分化受到抑制,成年新生颗粒神经元数量减少,而APN 干预(0.5 g/L)则能有效促进NSC增殖[33]。由此可见,APN能够参与促进海马神经发生。APN可激活p38有丝分裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)/GSK-3β/β 连环蛋白(β-catenin)信号通路,以剂量和时间依赖性的方式促进海马NSC 增殖[34]。对雄性小鼠进行AdipoRon(一种AdipoR1和AdipoR2 激动剂)干预后的结果显示,低剂量(20 mg/kg)的AdipoRon干预可提高血清脑源性神经营养因子(brain-derived neurotrophic factor,BDNF)水平,促进海马神经元增殖,而高剂量(50 mg/kg)的AdipoRon干预则抑制海马神经元增殖、分化与存活,损害小鼠空间识别记忆能力[35]。在另一项研究中,Song 等[36]发现,APN 干预(30 mg/L)能够逆转高糖环境对NSC 内AdipoR1表达的抑制,维持NSC 的神经球大小,促进NSC增殖与分化。综上可推断,适宜剂量的APN 能够对糖尿病相关的海马神经发生受损起到保护作用。
LEP也可调节海马神经发生。研究表明,LEP干预(1 mg/kg)能促进成年和老年小鼠海马DG和侧脑室室下区的NSC 增殖,促进新生神经元的增殖与分化[37]。Garza 等[38]研究证实,LEP 干 预(1 mg/kg)对于小鼠海马DG内NSC增殖的促进作用与PI3K/AKT 和JAK/STAT3 信号通路的活化有关。HFD 诱导人胰岛淀粉样多肽(human islet amyloid polypeptide,hIAPP)在胰岛内累积能促进T2DM发展。研究发现,HFD诱导的hIAPP转基因小鼠海马内胰淀素累积显著增加,且社会认知能力和被动学习能力显著下降[39]。高浓度(50~100 µg/L)的胰淀素抑制神经元增殖,而LEP 干预(10~100 µg/L)通过激活STAT3/腺苷酸活化蛋白激酶(adenosine 5'-monophosphate-activated protein kinase,AMPK)/ERK信号通路,逆转过量胰淀素对海马神经发生的抑制作用[40]。由此,LEP 可能缓解胰淀素累积导致的海马神经发生受损。
2.2 突触可塑性
突触可塑性(synaptic plasticity)是高级认知功能的神经结构基础,而高糖环境能损害海马突触可塑性,结构方面主要表现为突触结构损伤,功能方面主要表现为长时程增强(long-term potentiation,LTP)异常,源于N-甲基-D-天冬氨酸受 体(N-methyl-d-aspartate receptor,NMDAR)、α-氨基-3-羟基-5-甲基-4-异恶唑丙酸受体(α-amino-3-hydroxy-5-methyl-4-isoxazole-propionate receptor,AMPAR)和钾离子通道等功能紊乱[41]。对KKay 小鼠(一种T2DM 小鼠模型)的研究发现,其认知缺陷和LTP异常可能是由突触后谷氨酸能受体亚基的异常表达或磷酸化导致的[42]。该动物模型NMDAR 的NR1、NR2A 和NR2B 亚基的表达水平无异常,但NR2A和NR2B亚基的酪氨酸依赖性磷酸化水平显著降低,介导该过程的p-Src 表达水平降低,α 钙/钙调素依赖性蛋白激酶II(calcium/calmodulin-dependent protein kinase II α,CaMKIIα) 自 身 磷 酸 化 水 平 降 低,AMPAR 的GluR1亚基减少,GluR2亚基显著增加[42]。作为一种多功能的胰岛素增敏剂,APN 能有效增强啮齿类动物海马DG 高频刺激后的LTP,抑制低频刺激诱导的长时程抑制[43]。此外,从结构上看,APN-/-小鼠颗粒神经元的树突长度、分支和棘密度较对照组显著减少,而侧脑室注射APN(0.5 g/L)能增加新生颗粒神经元的树突棘和树突复杂性[44],从功能上看,APN-/-小鼠海马Schaefer 侧支通路的基础传递效能显著降低,突触前谷氨酸释放效率增加,谷氨酸能受体AMPAR 的GluA1 亚基以及NMDAR 的GluN1 和GluN2A 亚基水平减少,LTP 受损,AdipoR1/AMPK/GSK-3β/环磷腺苷效应元件结合蛋白 (cAMP-response element binding protein,CREB)信号通路活性受到抑制,同时空间学习和识别记忆能力显著降低[21]。Song 等[36]研究发现,HFD可抑制小鼠皮层、纹状体和海马内的AdipoR1和突触后致密蛋白95(postsynaptic density protein 95,PSD-95)以及皮层和海马内DCX 表达,导致突触功能障碍,而APN干预(30 mg/L)能通过维持AdipoR1的正常表达,保护突触免受脑内高糖环境的侵害。
研究发现,高水平的LepR 在海马的兴奋性突触处表达,且根据其在突触的定位,LEP能够调节SC-CA1 和TA-CA1 突触可塑性。LEP 缺乏或不敏感的啮齿类动物出现海马突触可塑性障碍和认知缺陷,而LEP干预能增强NMDAR功能,将海马短期增强转化为LTP,提高其记忆功能[44]。Hamilton等[45]认为,在不同生命阶段,LEP 对SC-CA1 和TA-CA1 突触发挥双重调控作用。对于SC-CA1 突触,在幼年期,LEP干预能够导致突触传递出现短暂或者持续的抑制,该过程涉及GluN2B 亚基和ERK信号的活化,在成年期,LEP通过选择性激活含有GluN2A 的NMDAR,诱导NMDA 依赖性LTP。该过程涉及蛋白酪氨酸磷酸酶的活性抑制,三磷酸磷脂酰肌醇表达水平升高以及缺乏GluA2亚基的AMPAR 的突触插入。对于TA-CA1 突触,在幼年期,LEP 干预能够通过刺激NMDA 受体亚基GluN2B, 并 涉 及PI3K 驱 动 的 缺 乏GluA2 的AMPAR 的突触插入诱导LTP,在成年期,LEP 干预依赖于GluN2A、JAK2/STAT3 信号传导以及从突触中去除缺乏GluA2 的AMPAR 诱导LTP。事实上,早在2001 年,便有研究认为,LEP 可通过激活PI3K、MAPK和Src酪氨酸激酶增强NMDAR介导的Ca2+流入,提高突触可塑性,且该过程的紊乱可能与糖尿病认知功能障碍的发生有关[46]。后续在糖尿病模型中进一步验证LEP在调节突触功能中的作用及机制对于理解糖尿病与认知功能障碍间的关系具有重要意义。
2.3 神经炎症
神经炎症(neuroinflammation)也是糖尿病诱发认知功能障碍的机制之一。研究表明,T2DM小鼠的认知缺陷与其海马内IL-6、IL-1β 和肿瘤坏死因子-α(tumor necrosis factor α,TNF-α)等促炎细胞因子的表达水平升高有关。高糖环境可通过活化哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)/核 因 子-κB (nuclear factor kappa B,NF-κB)信号通路,上调HT-22海马神经元IL-6、IL-1β和TNF-α 表达[47]。不仅如此,高糖环境下小胶质细胞的过度激活和星形胶质细胞增生也是诱发神经炎症的重要途径[48-49]。而APN 可能通过调节小胶质细胞和星形胶质细胞的生理活性,进而调控神经炎症。与对照组相比,APN-/-小鼠海马内小胶质细胞/巨噬细胞过度活化可诱发神经炎症和海马依赖性认知缺陷,而AdipoRon 干预(5 mg/kg)可激活过氧化物酶体增殖物激活受体γ(peroxisome proliferators-activated receptor γ,PPARγ)信号,逆转这些不良后果[50]。APN 也可缓解脂多糖(lipopolysaccharide,LPS)诱导的小胶质细胞炎症反应,该过程涉及AdipoR1/NF-κB和A20/Nod 样受体家族含pyrin 结构域蛋白3/半胱氨酸蛋白酶1(Caspase-1)等信号通路的活化或抑制[51-52]。此外,APN预处理(10 mg/L)能减少Aβ寡聚体诱导的BV2细胞IL-1β和TNF-α释放,恢复AMPK 磷酸化水平,抑制NF-κB 核转位。敲除AdipoR1 而非AdipoR2,可致该生理效应消失[53]。由此可见,APN 介导的小胶质细胞抗炎反应至少部分由AdipoR1 介导。在另一项研究中,APN 转染的内皮祖细胞干预能够限制星形胶质细胞活化,下调IL-6、IL-1β和TNF-α等表达,改善D-gal诱导的认知缺陷[54]。由此可见,基于APN基因修饰的细胞疗法也可能是缓解神经炎症以改善认知功能的有效方法。
LepR 也在小胶质细胞和星形胶质细胞表达,研究发现,LepR-/-大鼠脑内小胶质细胞的活化程度和吞噬能力较对照组显著增强,且在LPS 刺激下IL-6、IL-1β 和诱导型一氧化氮合酶(inducible nitric oxide synthase,iNOS)的分泌量显著增加,该生理现象与PI3K/AKT 信号通路的活性抑制有关[55]。由此可见,LepR信号的紊乱与神经炎症导致的认知功能障碍有关。张博等[56-57]研究表明,LEP在神经炎症中也可能发挥双向调节作用。正常生理状态下,过量LEP可激活JAK2/STAT3信号通路,促进BV2 细胞炎症反应;而在LPS 诱导下,适量的LEP 干预又可缓解小胶质细胞炎症反应。此外,在星形胶质细胞中,LPS 诱导LepR 表达增加,使IL-1β、环氧化酶2、Toll 样受体4 和胶质纤维酸性蛋白(glial fibrillary acidic protein,GFAP)含量增多,表明LEP 和LepR 可能调节星形胶质细胞的活化与增生[58]。Koga 等[59]对HFD 诱导的肥胖PS19 小鼠进行的研究结果显示,肥胖引起的高瘦素血症可导致星形胶质细胞活化、LEP超敏反应和LepR 表达水平过高,以加速PS19 小鼠的tau 病理。综上可知,LEP对小胶质细胞和星形胶质细胞的调控作用具有复杂性,且T2DM相关的高LEP水平可能诱发神经炎症。
2.4 氧化应激
高糖自氧化能够引起自由基过度产生,诱发氧化应激,损害脑组织结构,进而导致认知缺陷。对糖尿病前期和T2DM患者进行磁共振波谱检查的结果显示,血糖和糖化血红蛋白水平的升高与前扣带回谷胱甘肽(L-glutathione,GSH)水平降低有关[60]。此外,T2DM 患者体内氧化应激标志物8-异前列腺素F2α的水平升高与其精神灵活性和注意力的降低呈相关关系[61]。在动物研究中,王春等[62]表明,海马组织的氧化应激是诱发糖尿病认知功能障碍的重要因素。糖尿病大鼠的海马内脂质过氧化物(lipid peroxidation,LPO)增加,超氧化物歧化酶(superoxide dismutase,SOD)的活性降低,以及亚硝酸盐的水平升高[49]。
研究表明,长期HFD 可通过抑制神经元内AdipoR1/AMPK信号的活化,提高小鼠脑内活性氧(reactive oxygen species,ROS)/LPO 水平,减少GSH 含量,降低GSH/氧化型谷胱甘肽(GSSH)比值,进而增强氧化应激并导致认知缺陷。对胚胎小鼠海马细胞系mHippoE-14 进行的体外实验结果显示,AdipoR1 基因缺失可导致ROS/LPO 水平升高,核因子E2 相关因子2 (nuclear factor E2-related factor 2,Nrf-2)和血红素加氧酶1(heme oxygenase 1,HO-1)的蛋白质表达水平降低[63]。由此可见,APN 及其受体相关信号可能与海马神经元氧化应激有关。近年在氧和葡萄糖剥夺诱导的海马HT22神经元[64]、Sw-APP转染的SH-SY5Y细胞[65]和红藻氨酸诱导的海马神经元[66]中均已证实APN 具有抗氧化效应,然而APN 能否缓解高糖环境诱导的氧化应激需进一步验证。另有研究表明,在高糖环境中,PC12 细胞内ROS 和丙二醛(malonaldehyde,MDA)的水平显著升高,而LEP干预(12或24 nmol/L)可降低ROS和MDA水平,增加GSH含量,缓解高糖环境PC12细胞的氧化损伤[67]。由此可见,适量的LEP 能够保护高糖环境下氧化应激诱导的神经损伤。
2.5 神经元凋亡
高糖环境诱导的神经元凋亡(neuronal apoptosis)是糖尿病神经病变的重要原因之一。研究利用原代海马神经元、Neuro-2a 和SH-SY5Y 等多种细胞进行的实验结果证实,高糖环境能够诱导神经元凋亡[68-69]。Shi 等[69]研究表明,沉默信息调节因子2 相关酶1(silent mating type information regulation 2 homolog 1,SIRT1)的表达抑制以及p53乙酰化过度增加等信号的异常变化能够介导神经元凋亡,促进糖尿病认知功能障碍。
研究发现,APN 能够减轻糖尿病小鼠脑出血后的神经元凋亡、神经功能缺损和脑水肿。在体外高糖环境中,AdipoR1 活化后通过激活p53/p21 和c-Myc 信号调节NSC 存活[36]。APN 不仅以Smad3依赖性方式促进过氧化物酶体增殖物激活受体γ共激 活 因 子1α (peroxisome proliferator-activated receptor γ coactivator 1α,PGC-1α)相关的线粒体功能恢复,限制激活转录因子4 (activating transcription factor 4,ATF4)-CCAAT/增强子结合蛋 白 同 源 蛋 白 (enhancer-binding protein homologous protein,CHOP)信号诱导的神经元凋亡[70],也能够激活SIRT1/PGC-1α信号通路,上调B淋巴细胞瘤2(B-cell lymphoma 2,Bcl-2)表达,下调剪切型Caspase-3(Cleaved-Caspase-3)和Bax表达,进而抑制神经元凋亡[65]。此外,电针对糖尿病小鼠脑缺血损伤的改善也与APN 有关。APN可通过促进神经元AdipoR1 介导的GSK-3β 磷酸化,从而对糖尿病小鼠脑缺血再灌注损伤发挥保护作用[71]。另有研究报道,LEP 也参与调节神经元凋亡,LEP 能够以时间和剂量依赖性的方式激活JAK/STAT、PI3K 和MAPK 信号,下调凋亡因子Caspase-10和TNF相关的凋亡诱导配体表达,以抑制神经元凋亡[72]。在高糖环境诱导的PC12 细胞中,LEP 干预(12 或24 nmol/L)可抑制细胞毒性和Caspase-3 激活,降低Bax/Bcl-2 比值,缓解PC12细胞损伤[67]。深入探索LEP在改善糖尿病相关的神经元凋亡中的作用对于防治糖尿病认知功能障碍具有重要性和必要性。
3 运动介导脂肪因子改善糖尿病认知功能障碍的潜在机制
研究认为,缺乏运动是导致糖尿病认知功能障碍的危险因素之一[73],而适度运动能够改善糖尿病动物模型和患者的认知功能[74-75]。主要机制包括运动激活胰岛素信号、降低炎症、减少神经元凋亡和减轻血管损伤等[76]。此外,运动还是调控脂肪功能的重要方式,不仅能有效调节脂肪细胞内的转录因子表达、线粒体生物发生、葡萄糖转运和巨噬细胞浸润等,调控脂质分解、产热、胰岛素抵抗和炎症反应[77],也能调节脂肪组织的分泌功能,影响脂肪因子的合成与释放[78]。值得关注的是,运动可能介导脂肪因子改善糖尿病认知功能障碍。
3.1 运动、脂联素与认知功能
3.1.1 运动对脂联素合成与分泌的影响
n-3 多不饱和脂肪酸膳食摄入、噻唑烷二酮类给药和运动等干预方式能激活PPARγ 和AMPK 等信号并调节翻译后修饰、脂肪组织形态和巨噬细胞浸润等,增加APN合成与释放[79]。其中,运动促进APN合成与分泌的可能机制包括以下几个方面。a. 运动激活PPARγ 信号。PPARγ 是调控脂肪细胞分化的重要转录因子之一,也参与调节APN 表达[80]。陈霓等[81]研究发现,有氧运动可通过调节PPARγ 下游的APN 和TNF-α 表达,间接调控脂肪组织对胰岛素的敏感性。夏书宇等[82]研究报道,适宜强度的跑台运动能够改善肥胖大鼠脂肪代谢,上调脂肪组织PPARγ 和APN 的mRNA 表达,下调TNF-α 表达,提高血清APN 水平。因此,PPARγ可能是运动影响脂肪组织APN 合成与分泌的关键信号。b. 运动对“肝脏-脂肪”轴的调节作用。成纤维细胞生长因子21(fibroblast growth factor 21,FGF-21)是机体内重要的肝脏因子之一,通过与FGF 受 体(FGF receptor,FGFR) 和 辅 助 受 体β-klotho(KLB)结合形成受体复合物发挥生物学功能[83]。有研究发现,FGF-21干预能有效刺激脂肪组织分泌APN,且FGF-21 对糖尿病和肥胖小鼠血糖和胰岛素敏感性的影响由APN 介导[84-85]。运动可预防HFD 诱发的FGF-21-APN 信号损伤,增强FGF-21促进APN分泌的能力,提高肥胖小鼠血清APN 水平,改善糖代谢[86]。此外,PPARγ 信号也可能在FGF-21-APN 轴中发挥有效的调节作用[87]。长期HFD能够抑制小鼠脂肪组织FGFR1和KLB 表达,导致APN 生成过少以及脂肪酸释放过多,而跑台运动可通过PPARγ 介导的转录激活上调脂肪组织FGFR1和KLB表达,增强FGF-21信号的活性[88]。c. 运动降低脂肪组织炎症。研究发现,肥胖个体脂肪组织巨噬细胞(adipose tissue macrophages,ATMs)异常累积,表现为促炎的M1型ATMs增加,抗炎的M2型ATMs减少。脂肪组织炎症致使APN 表达和分泌量显著减少,而适度运动能减轻肥胖导致的脂肪组织炎症,减少M1型ATMs 并增加M2 型ATMs,促进APN 分泌且抑制IL-6 和TNF-α 分泌[89]。综上,运动促进脂肪组织APN合成与分泌的作用可能与PPARγ信号激活、FGF-21-APN轴活化以及脂肪组织炎症水平降低有关(图2)。
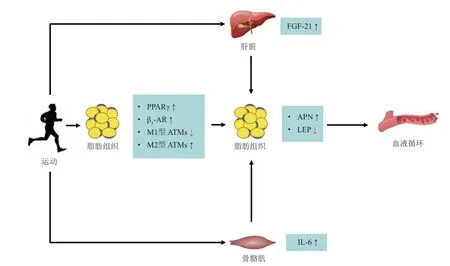
Fig. 2 Mechanism of exercise regulating adiponectin and leptin secretion in adipose tissue图2 运动调控脂肪组织脂联素和瘦素分泌的机制
3.1.2 运动介导脂联素改善糖尿病认知功能障碍的潜在作用
综合表1 中研究结果发现,持续8~12 周、每周3~5次、每次20 min以上的有氧运动(以跑台运动为主)能显著提高糖尿病动物模型体内的APN水平,且有氧运动的效果要优于抗阻运动或者联合运动[90]。除调节机体能量代谢外,有氧运动也可介导APN改善啮齿类动物的空间学习和记忆能力。具体而言,有氧运动能增加小鼠海马组织APN 含量,激活APN-Notch 信号通路,促进海马神经发生,改善空间学习和记忆能力[91-93]。与对照组相比,糖尿病小鼠海马APN 水平显著降低,成年海马神经发生受到抑制。为期2周的自主跑轮运动能有效促进糖尿病小鼠海马神经发生,但对于APN缺乏的小鼠却无此效应[94]。由此可见,运动对糖尿病动物模型海马神经发生的促进作用至少部分是由APN 及相关信号介导的。此外,Lee 等[95]研究证实,慢性AdipoRon 干预(20 mg/kg)能够模拟自主跑轮运动对于海马神经可塑性的提高效应,促进糖尿病小鼠海马DG内NSC增殖和分化,也可通过激活AMPK/PGC-1α 信号通路,提高海马DG 的树突复杂性、棘密度和NMDAR依赖性LTP,增加BDNF含量,增强小鼠空间识别记忆能力。综上所述,运动介导APN 改善糖尿病动物模型的认知功能与海马神经发生和突触可塑性的增强有关。
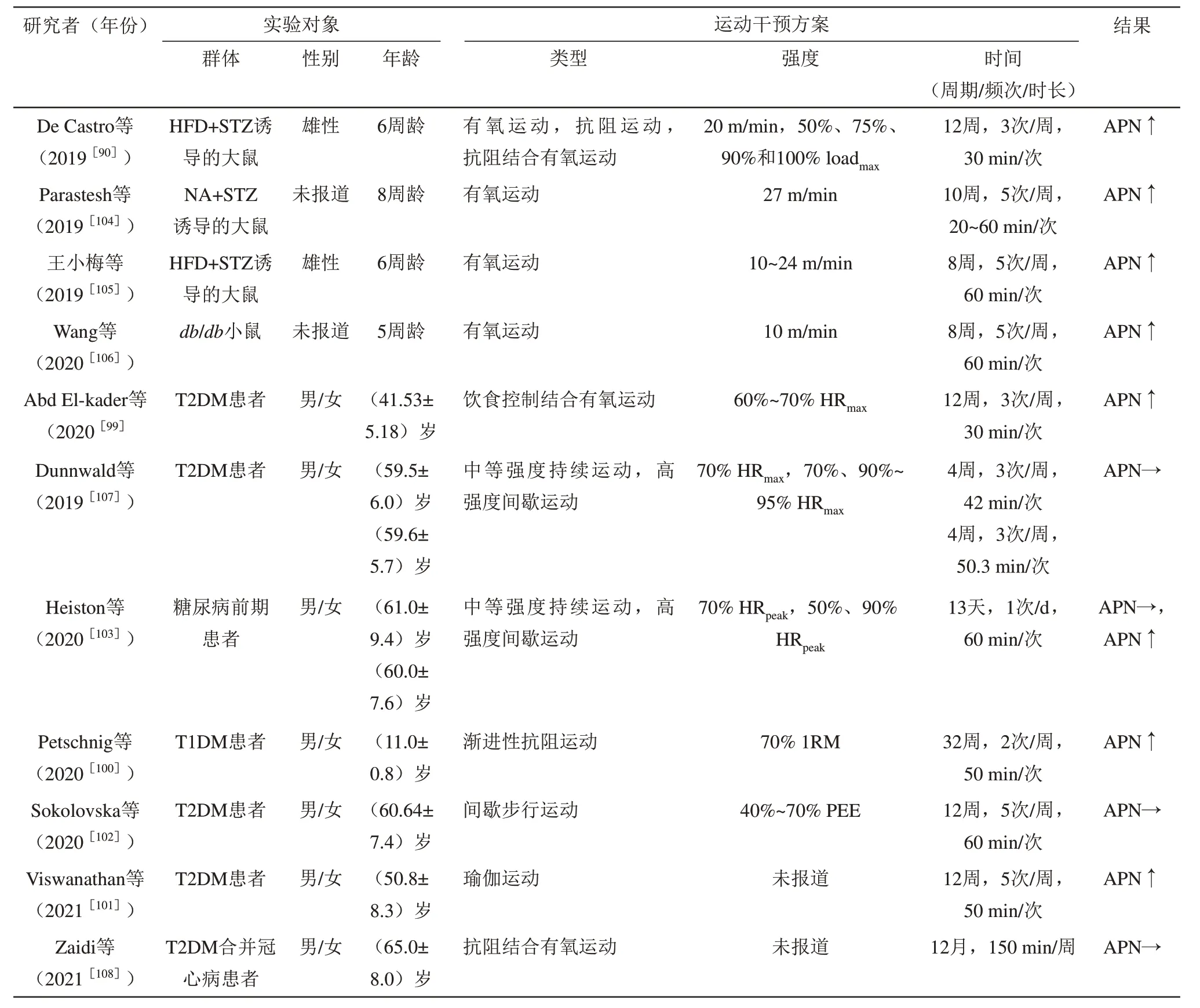
Table 1 Effects of exercise on adiponectin levels in diabetic animal models and patients表1 运动对糖尿病动物模型和患者脂联素水平的影响
人体研究表明,运动对认知功能的改善作用也与APN 的水平升高有关。针对瘦体型年轻受试者的研究发现,急性高强度(75%~80% HRmax)有氧运动能够增强大脑对脑脊液APN 的摄取作用,提高大脑可塑性[96]。对于老年糖尿病患者而言,为期3 个月、每周5 次、每次40~60 min 的有氧运动能显著提高老年糖尿病合并MCI患者的认知功能,尤其是延迟记忆、视空间能力和执行功能等,且该效应与血清APN 的水平升高,血糖、血脂以及胰岛素抵抗的水平降低有关[97]。此外,韦忠培等[98]研究表明,为期3个月、每周5次、每次40~60 min的低至中等强度(50%~70% HRmax)有氧运动可显著提高老年糖尿病合并MCI 患者血清APN 的水平以及视空间能力、执行功能、注意力、语言和延迟记忆等功能。由此,运动可能通过提高APN 水平改善老年糖尿病合并MCI患者的认知功能。然而,由于受试者异质性以及运动干预要素的差异,不同研究中运动对糖尿病患者体内APN 水平的影响出现差异结果。综合表1中研究结果发现,尽管规律性的有氧运动、抗阻运动和瑜伽等不同模式的运动干预均能提高糖尿病患者体内APN 水平[99-101],但运动强度可能影响运动效果。低至中等强度的间歇性步行运动对糖尿病患者体内APN 水平的影响并不显著[102]。此外,在Heiston等[103]的研究中,仅高强度间歇运动而非中等强度持续运动能显著提高糖尿病前期患者血清APN 的水平。因此,强度可能是运动影响糖尿病患者体内APN 水平重点关注的要素之一。
3.2 运动、瘦素与认知功能
3.2.1 运动对瘦素合成与分泌的影响
研究表明,脂肪组织LEP 合成与分泌受运动和碳水化合物补充等干预的影响[109]。长期缺乏运动可导致个体血浆LEP的浓度显著增加[110],而适度运动则能降低循环LEP 的水平[111]。运动影响LEP 合成与分泌的可能机制体现在以下几个方面。a. 运动激活β 肾上腺素能受体(β-adrenergic receptor,β-AR)。研究认为,白色脂肪细胞LEP的合成受交感神经系统调节,且β-AR 参与介导这种调节作用[112]。其中,β3-AR 的激活不仅能够介导能量消耗、棕色脂肪产热和白色脂肪分解,也抑制LEP基因表达并降低血清LEP水平[113]。人体研究中,β3-AR基因多态性被证实与规律性运动对葡萄糖耐量和瘦素抵抗的改善作用有关[114]。动物研究也发现,急性运动能通过激活β3-AR下调雄性大鼠腹膜后脂肪组织LEP的mRNA表达[115]。b. 运动对“肌肉-脂肪”轴的调节作用。IL-6 是经典的肌肉因子之一。Brandt等[116]动物研究表明,运动能通过增加肌肉来源的IL-6 含量,调节小鼠皮下脂肪组织的代谢适应,下调脂肪组织LEP 的mRNA表达。c. 运动降低脂肪组织炎症。缺乏运动或肥胖影响脂肪组织的免疫代谢,导致血浆LEP 浓度显著增加[117]。而适度运动能有效抑制肝脏脂肪变性以及白色脂肪组织巨噬细胞浸润,促进IL-10 等抗炎细胞因子分泌,限制LEP 的分泌[118-119]。上述证据表明,运动对脂肪组织LEP 合成与分泌的影响与β3-AR信号激活、IL-6-LEP轴活化以及脂肪组织炎症水平降低有关(图2)。
3.2.2 运动介导瘦素改善糖尿病认知功能障碍的潜在作用
正常生理状态下,有氧运动能通过增加LEP的水平和LepR 的活性以发挥促认知效应。动物研究表明,母体在孕期进行有氧运动可上调子代小鼠海马LepR 表达,提高海马神经元存活率,同时行为学表现出空间学习和记忆能力增强[120]。在另一项研究中,低强度的有氧运动和/或天然抗氧化剂虾青素干预能够增加小鼠海马LEP 含量,激活AKT/STAT3 信号通路,促进海马神经发生,提高小鼠认知功能,且当两者联合时干预效果更佳[121]。Uysal等[122]研究表明,有氧运动可显著上调雄性大鼠皮层和海马内LEP、LepR 和胰岛素样生长因子1 (insulin-like growth factor 1,IGF-1)的表达,且LEP、LepR与IGF-1的水平呈显著正相关。综上可知,正常生理状态下的运动刺激可能通过增强LEP 相关信号提高个体认知。然而,对衰老F344 大鼠进行的研究发现,运动能够降低其皮质内的LEP 水平,促进海马神经发生,缓解神经炎症,改善衰老大鼠的认知缺陷[123]。在肥胖PS19小鼠中,高瘦素血症导致脑内LEP 信号紊乱,而自主跑轮运动可提高LEP 敏感性,缓解高胰岛素诱导的神经炎症[59]。因此,在体内LEP 的水平异常升高时,适度运动又可能通过降低LEP 水平并提高瘦素敏感性,以调节海马神经发生和神经炎症,进而改善认知功能。综合表2 中研究结果发现,持续4~8 周、每周3~5 次、每次30~60 min 的跑台运动或爬梯运动能够显著降低糖尿病大鼠体内LEP水平。然而李寿邦等[124]研究结果显示,游泳运动和/或魔芋多糖干预可提高糖尿病大鼠血清LEP水平。未来仍需对现有研究中矛盾结果出现的原因进行深入剖析。

Table 2 Effects of exercise on leptin levels in diabetic animal models and patients表2 运动对糖尿病动物模型和患者瘦素水平的影响
尽管人体研究证实,肥胖个体定期运动获得的促认知效应与血清LEP 的水平降低有关[125],然而,当前关于运动通过调节LEP 水平改善糖尿病患者认知功能的研究较为匮乏。此外,与APN 情况相似,不同研究中运动对糖尿病患者体内LEP水平的影响效果也存在差异。例如,Dunnwald等[107]研究发现,规律性的中等强度持续性运动和高强度间歇性运动均未显著改变糖尿病患者体内LEP 水平,这与Heiston 等[103]研究结果不一致。然而,在这两项研究中,由于受试者的病程以及运动干预的周期、频次、时间和强度等要素均不相同,因而难以明确结果不一致的原因。此外,尽管运动周期长达16 周,但体育舞蹈训练和间歇步行运动难以显著改变糖尿病患者体内LEP 水平[102,126]。因此,后续在控制不同变量的条件下进一步探索运动干预要素对糖尿病患者体内LEP 水平的影响对于运动干预方案的优化与应用至关重要。
4 总结与展望
随着生活方式的改变,糖尿病和肥胖等代谢综合征成为认知功能障碍的重要诱因。研究表明,APN 和LEP 等脂肪因子对糖尿病认知功能障碍的早期诊断与干预具有重要价值。APN 和LEP 不仅能通过调节血糖和胰岛素敏感性间接影响个体认知功能,也可穿过BBB进入大脑,通过结合其受体,激活或抑制神经元或神经胶质细胞中的p38 MAPK、AMPK、ERK、JAK2/STAT3、PI3K/AKT和SIRT1/PGC-1α 等信号通路,以调节神经发生、突触可塑性、神经炎症、氧化应激和神经元凋亡等进程,进而调控认知功能(图1)。此外,APN 和LEP还可作为运动改善糖尿病认知功能障碍的重要介质。对于糖尿病动物模型和糖尿病患者而言,运动可能通过提高循环APN 的水平,降低LEP 水平并提高LEP敏感性,以促进海马神经发生,提高突触可塑性,降低神经炎症,从而改善认知功能。
相关领域的后续研究可致力于解决如下问题:a. 虽然大量研究已证实,维持体内适宜浓度的APN 和LEP 可能是改善糖尿病认知功能障碍的关键,但其间的“剂量-效应”关系尚未完全建立;b. 除APN 和LEP 外,近年研究发现,其他脂肪因子如鸢尾素、爱帕琳肽、抵抗素和脂质运载蛋白2等也可能与糖尿病认知功能障碍之间存在紧密关联[131-135],但具体关系及其内在机制仍有待进一步揭示;c. 未来关于运动介导脂肪因子改善糖尿病认知功能障碍的最佳类型、强度、频次和周期等要素的确立,将有助于推动“运动+药物”精准医疗的发展。
猜你喜欢
作文周刊·小学二年级版(2022年20期)2022-05-05
中老年保健(2021年9期)2021-08-24
昆明医科大学学报(2021年8期)2021-08-13
创新作文(小学版)(2019年10期)2019-09-25
中国生殖健康(2019年12期)2019-01-07
小学生学习指导(低年级)(2017年5期)2017-05-04
分子影像学杂志(2015年3期)2015-12-04
西南军医(2015年3期)2015-04-23
河南医学研究(2014年2期)2014-02-27
食品科学(2013年15期)2013-03-11
