
氟西汀对Aβ25-35诱导小胶质细胞NLRP3炎性小体活化及细胞焦亡的影响
2023-06-10 10:47马晓涵韩滨高凡迟松
青岛大学学报(医学版) 2023年2期
马晓涵,韩滨,高凡,迟松
(青岛大学附属医院神经内科,山东 青岛 266003)
阿尔茨海默病(AD)是一种以进行性认知障碍、记忆减退和精神行为症状为主要临床特征的神经退行性疾病,其致残率高,病程较长,目前尚无有效的防治策略[1]。AD的发病机制存在多种假说,其中β-淀粉样蛋白(Aβ)激活小胶质细胞的炎症反应被认为在AD的发病过程中起着关键作用[2-3]。核苷酸结合寡聚化结构域样受体蛋白3(NLRP3)炎性小体是由NLRP3受体、凋亡相关斑点样蛋白(ASC)和半胱氨酸蛋白酶-1(caspase-1)组成的多蛋白复合体[4],在中枢神经系统中主要表达于小胶质细胞[5]。研究表明,NLRP3 炎性小体可被Aβ激活,进而促进Aβ的沉积和微管蛋白tau 的过度磷酸化[6-9]。此外,NLRP3 炎性小体的激活能够触发小胶质细胞的焦亡,这是一种新的程序性的细胞死亡形式,表现为细胞不断肿胀直至细胞膜破裂,释放大量炎性递质,加剧神经炎症反应[10]。
氟西汀是一种选择性的5-羟色胺再摄取抑制剂,在临床上广泛应用于抑郁症的治疗。最近的研究发现,氟西汀能够通过抗炎、抗氧化应激发挥神经保护作用[11-12]。在抑郁动物模型中,氟西汀能通过下调 NLRP3 信号通路抑制小胶质细胞的活化和炎性因子的分泌[13-14]。尽管研究发现抗抑郁药的长期应用能够降低AD 罹患风险并改善认知能力[15-17],但是氟西汀能否抑制AD环境下小胶质细胞焦亡和神经炎症目前尚无报道。本研究使用Aβ25-35刺激 BV2 小胶质细胞模拟AD炎性环境,以NLRP3炎性小体与细胞焦亡为切入点,探讨氟西汀对AD的保护作用及其潜在机制。
1 材料与方法
1.1 实验材料
永生系小鼠BV2小胶质细胞购于武汉普诺赛公司。氟西汀购于美国Sigma公司。Aβ25-35、双硫仑和CCK8检测试剂盒购于美国MCE公司;胎牛血清、青链霉素混合液以及MEM培养液购自武汉普诺赛公司;BCA蛋白检测试剂盒、Annexin V-FITC/PI 细胞凋亡检测试剂盒、白细胞介素1β(IL-1β)及白细胞介素18(IL-18)的酶联免疫吸附试验(ELISA)检测试剂盒、山羊抗兔二抗、山羊抗鼠二抗购自武汉伊莱瑞特公司;乳酸脱氢酶(LDH)检测试剂盒购自北京索莱宝公司;PBS缓冲液、RIPA裂解液和DCFH-DA活性氧(ROS)检测试剂盒购自上海碧云天公司;ECL试剂盒购于武汉博士德公司;caspase-1抗体购于美国Santa cruz公司;NLRP3、ASC和活化半胱氨酸蛋白酶-1(cleaved-caspase-1)抗体购于美国CST公司;GAPDH和膜穿孔蛋白D(GSDMD)抗体购自美国Proteintech公司。
1.2 实验方法
1.2.1细胞培养及分组 BV2细胞生长于含有体积分数0.10胎牛血清和10 g/L青链霉素的MEM培养液中,置于含体积分数0.05 CO2的37 ℃恒温培养箱内培养24 h后传代。本研究使用第3~5代的细胞。首先将BV2细胞以每孔大约5×103个细胞的密度种植于96孔板中,用不同浓度的Aβ25-35处理12、24、36、48 h,利用CCK8检测细胞活力,获得Aβ25-35最佳处理浓度(20 μmol/L)和时间(24 h)。实验分为以下6组:空白对照组(A组),Aβ25-35组(B组),Aβ25-35+小剂量(5 μmol/L)氟西汀组(C组),Aβ25-35+中剂量(浓度10 μmol/L)氟西汀组(D组),Aβ25-35+高剂量(浓度20 μmol/L)氟西汀组(E组),Aβ25-35+双硫仑(10 μmol/L)组(F组)。除空白对照组外,其他各组用不同浓度的氟西汀或双硫仑预处理BV2细胞2 h,再加入20 μmol/L的Aβ25-35培养24 h用于后续实验。
1.2.2CCK8法检测细胞活力 将BV2细胞以大约每孔5×103个细胞的密度接种到96孔板中,待其均匀贴壁生长后,用不同浓度的Aβ25-35、氟西汀、双硫仑进行处理。然后在规定的不同时间点每孔加入10 μL CCK8试剂,在37 ℃下孵育2.5 h。最后用酶标仪测量各组细胞在450 nm波长下的吸光度(A)值,细胞存活率=(实验孔A值-空白孔A值)/(对照孔A值-空白孔A值)×100%。
1.2.3ELISA法检测细胞上清液中IL-1β和IL-18含量 收集处理好的各组细胞上清液,根据ELISA试剂盒说明书的步骤测定IL-1β和IL-18水平。
1.2.4LDH释放量检测 LDH的释放反映了细胞质膜完整性的丧失,可间接反映细胞焦亡的程度。收集各组细胞上清液,根据LDH试剂盒说明步骤测定LDH的释放量。
1.2.5Annexin V-FITC/PI染色 早期凋亡细胞被Annexin V标记,而焦亡和中晚期凋亡细胞可被Annexin V和PI同时标记,所以Annexin V/PI双阳性率可作为细胞焦亡比例的间接指标。收集各组细胞后按Annexin V-FITC/PI双染色凋亡检测试剂盒说明步骤处理,上流式细胞仪检测各组细胞Annexin V/PI双阳性率,使用Flowjo分析数据。
1.2.6ROS生成的检测 按照DCFH-DA试剂盒说明步骤处理各组细胞,使用Image J 6.0分析各组荧光强度(细胞内的ROS可以氧化无荧光的DCFH生成有荧光的DCF,ROS含量与荧光显微镜下测量的DCF荧光强度呈正比)。
1.2.7Western blot法检测炎性小体及焦亡相关蛋白表达 处理好的各组细胞以冷PBS洗涤3次后用细胞刮刀刮下置于离心管中,加入RIPA裂解液冰上裂解30 min。细胞裂解物于4 ℃下以12 000 r/min离心20 min,取上清(即蛋白样品)。使用BCA蛋白测定试剂盒测定蛋白浓度。各组蛋白样品加入5×上样缓冲液,金属浴100 ℃煮5 min使蛋白变性。煮好的蛋白按照每孔20 μg上样,进行SDS-PAGE电泳并湿转到PVDF膜上,再用体积分数0.05的脱脂牛奶室温封闭1.5 h,分别加入NLRP3(1∶1 000)、ASC(1∶1 000)、caspase-1(1∶500)、cleaved-caspase-1(1∶1 000)、GSDMD(1∶5 000)、GAPDH(1∶5 000)抗体,4 ℃过夜。以TBST溶液洗膜3次,分别加入稀释好的山羊抗兔IgG(1∶5 000)或山羊抗鼠IgG(1∶5 000)室温孵育1.5 h。以TBST溶液洗膜3次,ECL发光液显影,扫描后用Image J 6.0分析相应条带灰度值,以GAPDH为内参,计算各目标蛋白的相对表达量。
1.2.8电镜检测 各组处理好的细胞以冷PBS洗涤2次后用细胞刮刀刮下置于离心管中进行离心(1 000 r/min,5 min),弃去上清液,将离心管中的细胞团块置于体积分数0.025的戊二醛固定液(pH 7.3~7.4)中,4 ℃保存,最后用透射电子显微镜观察细胞形态变化。
1.3 统计学分析

2 结 果
2.1 氟西汀对Aβ25-35诱导的BV2细胞活力的影响
析因设计的方差分析结果显示,Aβ25-35的作用浓度与作用时间都对细胞存活率存在显著影响,Aβ25-35在10~40 μmol/L浓度范围内处理BV2细胞24 h后,细胞活力均受到明显的抑制,其中应用20 μmol/L Aβ25-35时细胞的存活率接近50%,表明该浓度可对细胞造成有效伤害,因此选择24 h和20 μmol/L作为Aβ25-35的干预时间和干预浓度。见表1。不同浓度氟西汀处理2 h后的细胞活力差异具有统计学意义(F=11.202,P<0.05),组间两两比较,与空白对照组相比,1~20 μmol/L氟西汀对细胞活力影响不明显(P>0.05),40和60 μmol/L氟西汀则显著降低了细胞活力(P<0.05)(图1a),因此后续实验中选择5、10和20 μmol/L作为氟西汀的治疗浓度。空白对照组、Aβ25-35组和不同浓度氟西汀预处理组的细胞活力差异具有统计学意义(F=19.664,P<0.05),两两比较结果显示,10和20 μmol/L氟西汀预处理细胞2 h逆转了Aβ25-35诱导的细胞活力下降(P<0.05)(图1b)。
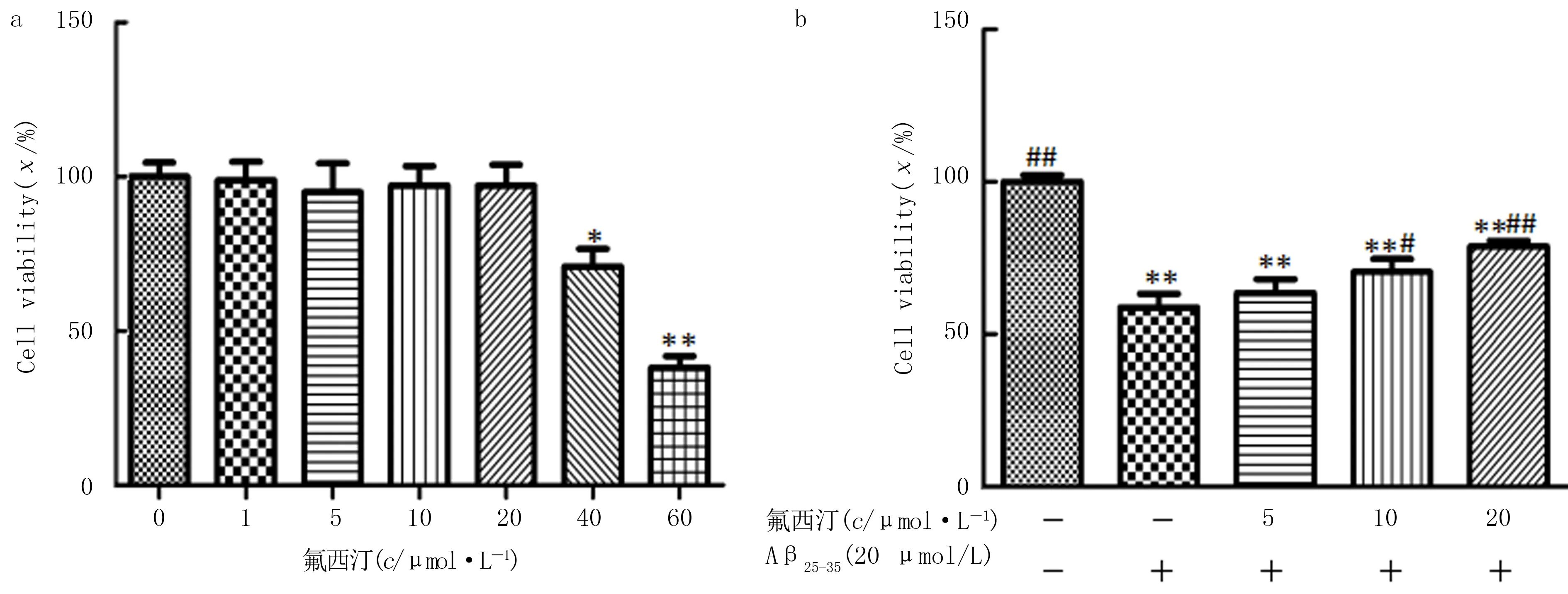
a:氟西汀对BV2细胞活力的影响(n=6);b:氟西汀对Aβ25-35诱导的BV2细胞活力的影响(n=5)。与空白对照组比较,*P<0.05,**P<0.01;与Aβ25-35组比较,#P<0.05,##P<0.01。图1 氟西汀对Aβ25-35诱导的BV2细胞活力的影响
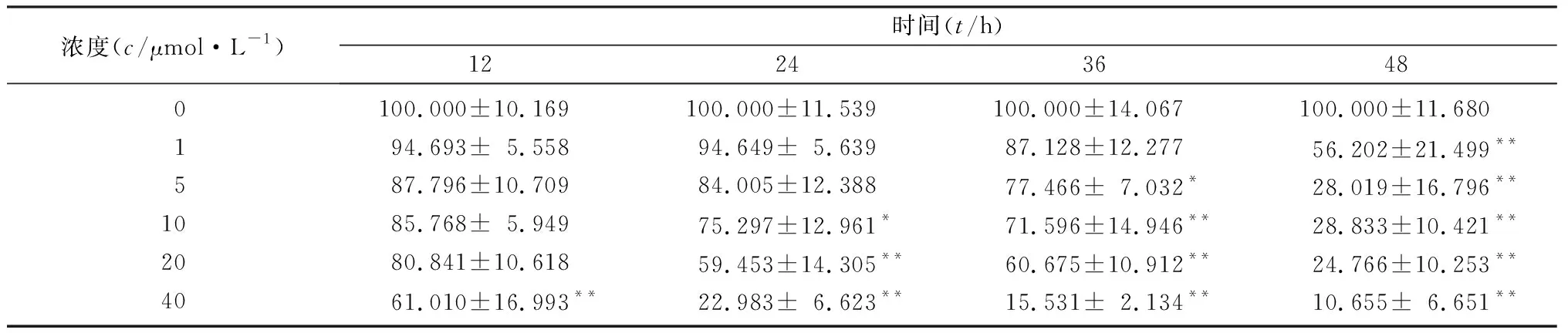
表1 不同浓度Aβ25-35作用不同时间对BV2细胞存活率的影响(n=30,χ/%)
2.2 氟西汀对Aβ25-35诱导的BV2细胞焦亡的影响
Annexin V-FITC/PI染色结果显示,20 μmol/L Aβ25-35显著增加了PI阳性的BV2细胞数量,而不同浓度的氟西汀预处理降低了PI阳性的细胞数量,且呈剂量依赖性(图2a)。PI阳性细胞的定量分析显示,各处理组间焦亡程度差异具有统计学意义(F=35.938,P<0.01),两两比较结果显示,高剂量(20 μmol/L)的氟西汀能够显著降低细胞焦亡比例(P<0.01), 但中低剂量的氟西汀对Aβ25-35诱导的细胞焦亡抑制不显著(P>0.05)(图2b)。各处理组间LDH释放量差异也具有统计学意义(F=6.172,P<0.05),组间两两比较结果显示,10和20 μmol/L氟西汀预处理降低了Aβ25-35诱导的BV2细胞中LDH的释放(P<0.05)(图2c)。在形态学上,利用透射电镜可观察到20 μmol/L Aβ25-35刺激后的BV2细胞出现肿胀变形、细胞膜完整性破坏及大量焦亡小体(空泡变性)等典型的细胞焦亡特征;与Aβ25-35组相比,20 μmol/L氟西汀处理组细胞焦亡缓解,细胞形态变化不明显(图2d)。以上结果表明,氟西汀的应用,尤其是高剂量(20 μmol/L)氟西汀可以有效抑制Aβ25-35诱导的BV2细胞焦亡。

a:Annexin V-FITC/PI双染色法检测细胞焦亡;b:PI阳性细胞定量分析(n=3);c:各组LDH的释放量比较(n=4);d:透射电镜观察BV2细胞焦亡形态变化,红色箭头所指为焦亡小体。与空白对照组比较,*P<0.05,**P<0.01;与Aβ25-35组比较,#P<0.05,##P<0.01。图2 氟西汀对Aβ25-35诱导的BV2细胞焦亡的影响
2.3 氟西汀对BV2细胞中Aβ25-35诱导的GSDMD表达影响
各处理组间GSDMD蛋白的表达差异具有统计学意义(F=4.001,P<0.05);与空白对照组相比较,Aβ25-35组细胞中GSDMD的表达显著增加(P<0.01),而20 μmol/L氟西汀和10 μmol/L 双硫仑(GSDMD特异性拮抗剂)处理后GSDMD蛋白表达均较Aβ25-35组明显降低(P<0.05)(图3a)。此外,双硫仑对BV2细胞活力影响也具有统计学差异(F=44.778,P<0.01),其中10 μmol/L双硫仑能够逆转Aβ25-35诱导的BV2细胞活力下降(P<0.05)和细胞焦亡(图2a、3b~e)。
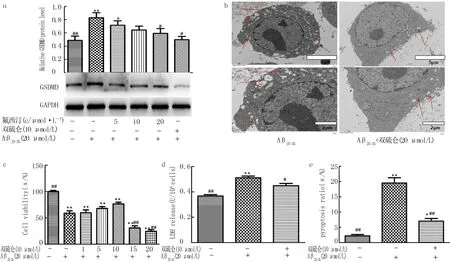
a:Western blot法检测GSDMD蛋白的表达(n=5);b:透射电镜观察BV2细胞形态变化;c:双硫仑对Aβ25-35诱导的BV2细胞活力的影响(n=5);d:各组LDH释放量的比较(n=4,F=8.137,P<0.05);e:PI阳性细胞定量分析(n=3,F=59.766,P<0.01)。与空白对照组相比较,*P<0.05,**P<0.01;与Aβ25-35组比较,#P<0.05,##P<0.01。图3 氟西汀对BV2细胞中Aβ25-35诱导的GSDMD表达影响
2.4 氟西汀对BV2细胞中Aβ25-35诱导的NLRP3炎性小体激活的影响
各处理组间的NLRP3(F=2.787,P<0.05)、ASC(F=5.832,P<0.05)、caspase-1(F=3.164,P<0.05)、cleaved-caspase-1(F=3.145,P<0.05)蛋白表达比较差异均具有统计学意义。两两比较结果显示,与空白对照组相比,Aβ25-35组上述炎性小体相关蛋白的表达均显著上调(P<0.05),但该上调作用在高剂量(20 μmol/L)氟西汀预处理后均得到了抑制(P<0.05)。见图4、表2。
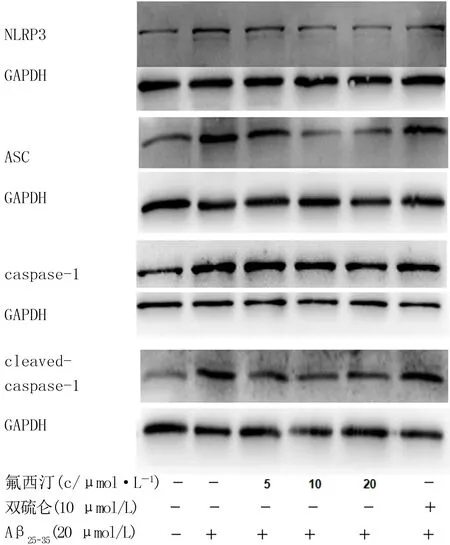
Western blot法检测NLRP3炎性小体相关蛋白的表达水平,以GAPDH为内参。图4 氟西汀对BV2细胞中Aβ25-35诱导的NLRP3炎性小体激活的影响

表2 各组细胞炎性小体和焦亡相关蛋白以及ROS产生的比较
2.5 氟西汀对炎性小体相关促炎因子表达影响
各组间IL-1β(F=9.189,P<0.05)、IL-18(F=6.680,P<0.05)表达比较差异均有显著性。两两比较结果显示,与空白对照组相比较,Aβ25-35组细胞上清液中IL-1β、IL-18的含量均显著增加(P<0.01),而不同浓度的氟西汀(5、10、20 μmol/L)和双硫仑(10 μmol/L)则抑制了IL-1β和IL-18的表达(P<0.05)。见表2。
2.6 氟西汀对Aβ25-35诱导的BV2细胞中ROS产生的影响
各处理组间ROS荧光强度比较差异具有显著性(F=15.159,P<0.05)。两两比较结果显示,与空白对照组相比,Aβ25-35组ROS荧光强度明显增强(P<0.01);而不同浓度的氟西汀均抑制了Aβ25-35诱导的ROS的生成,且呈剂量依赖性(P<0.05);但10 μmol/L双硫仑处理组与Aβ25-35组相比,差异无显著性(P>0.05)。见图5、表2。

荧光显微镜检测各组ROS的生成,荧光定量分析法测定ROS水平。
3 讨 论
研究发现,氟西汀能够在多种疾病中通过抑制NLRP3炎性小体活化进而减轻神经炎症反应并改善神经功能,如抑郁症[13]、脑损伤[18]和萎缩性黄斑变性[19]。本研究结果证实,Aβ25-35能够诱导BV2细胞NLRP3炎性小体的激活和细胞焦亡,促进炎性细胞因子IL-1β和IL-18的释放,而这些都可以被外源性的氟西汀所抑制,提示氟西汀可能通过抑制NLRP3炎性小体激活、小胶质细胞焦亡以及随后的炎症级联反应对AD发挥神经保护作用。越来越多的证据表明,NLRP3信号通路在AD的发生和发展中发挥着关键作用[20]。关于Aβ如何介导小胶质细胞NLRP3炎性小体激活目前已有多种假说被提出。有研究认为,Aβ作为NLRP3炎性小体的启动或活化刺激物,最终导致由caspase-1介导的炎性因子(IL-1β、IL-18)的释放[21],而且在特定条件下,活化的caspase-1将裂解下游的成孔蛋白GSDMD,从而促进小胶质细胞发生焦亡[22-23]。本实验结果表明,氟西汀的应用,尤其是高剂量(20 μmol/L)的氟西汀可以有效缓解Aβ25-35诱导的BV2细胞焦亡,主要表现为LDH释放量降低、PI阳性的BV2细胞数量减少以及细胞形态学上的变化;此外,氟西汀也可以逆转Aβ25-35诱导的细胞活力下降。GSDMD不仅是焦亡的关键因子,也是焦亡成孔的候选蛋白之一[24-25]。为了明确GSDMD是否在Aβ25-35诱导的细胞焦亡中发挥作用,本研究使用10 μmol/L双硫仑(GSDMD特异性拮抗剂)作为对照,实验结果显示,双硫仑有效抑制了GSDMD的表达和细胞焦亡,初步证实了GSDMD在Aβ25-35诱导的细胞焦亡中发挥关键作用。此外,高剂量(20 μmol/L)的氟西汀可以有效降低Aβ25-35诱导的BV2细胞GSDMD的表达进而改善焦亡。
本团队前期研究显示,氟西汀能够通过抑制NLRP3炎性小体和caspase-1的活化改善蛛网膜下隙出血和抑郁[13,18]。AMBATI等[19]的研究显示,氟西汀可直接与NLRP3蛋白结合并阻止NLRP3-ASC炎症小体的组装和激活,进而缓解萎缩性黄斑变性。本研究探讨了氟西汀是否可靶向抑制Aβ25-35介导的NLRP3炎性小体激活,结果显示,高剂量(20 μmol/L)氟西汀下调NLRP3炎性小体相关蛋白(NLRP3、ASC、caspase-1、cleaved-caspase-1)的表达,提示氟西汀可能通过抑制NLRP3炎性小体激活对AD产生保护作用。已知IL-1β和IL-18在脑内神经炎症的启动和维持中发挥关键作用[26]。与先前在抑郁模型中的研究结果一致[14],本研究结果表明,氟西汀的应用能显著抑制Aβ25-35诱导的IL-1β和IL-18产生。
ROS除了作为NLRP3炎性小体的重要上游活化信号外,还与AD的发生发展密切相关[27]。有研究结果表明,氟西汀能在多种情况下减少ROS的生成[28]。本文研究结果显示,与Aβ25-35组相比较,不同浓度的氟西汀预处理显著降低了细胞内ROS含量,且该作用呈剂量依赖性。由此推测,氟西汀可能通过抑制Aβ25-35诱导的ROS生成抑制NLRP3炎性小体的活化。
综上所述,氟西汀能够抑制Aβ25-35诱导的BV2小胶质细胞ROS产生、NLRP3炎性小体激活和细胞焦亡,进而改善神经炎症反应,氟西汀可能通过调节ROS/NLRP3/GSDMD信号通路在AD中发挥神经保护作用,这为延缓AD发展提供了新的治疗思路,值得进一步深入研究。
猜你喜欢
世界科学技术-中医药现代化(2022年3期)2022-08-22
医学综述(2022年7期)2022-04-19
医学概论(2022年1期)2022-03-22
昆明医科大学学报(2020年12期)2021-01-26
世界科学技术-中医药现代化(2020年2期)2020-07-25
药学与临床研究(2019年2期)2019-05-14
中国组织化学与细胞化学杂志(2017年1期)2017-06-15
西南军医(2016年1期)2016-01-23
吉林大学学报(医学版)(2015年1期)2015-12-17
中国当代医药(2015年30期)2015-03-01
