
一种基于线粒体分离纯化技术的新型线粒体DNA深度测序方法*
2023-06-04 11:30:04朱玉青朱琳黄明涛胡平沈彬梁栋许争峰南京医科大学附属妇产医院南京市妇幼保健院产前诊断中心南京0004南京医科大学生殖医学国家重点实验室南京66
临床检验杂志 2023年3期
朱玉青,朱琳,黄明涛,胡平,沈彬,梁栋,许争峰(.南京医科大学附属妇产医院&南京市妇幼保健院产前诊断中心,南京 0004;.南京医科大学生殖医学国家重点实验室,南京 66)
线粒体是真核细胞中负责能量代谢、氧化应激、细胞凋亡等重要功能的细胞器,拥有一套独立于细胞核基因组的遗传物质,即线粒体基因组(mtDNA)[1]。人线粒体基因组是1个长度为16 569 bp的环状双链DNA分子,以多拷贝的形式存在于细胞中。mtDNA由于缺少DNA损伤修复系统,故而较之细胞核基因组更易产生突变[2],并且mtDNA突变可通过母系遗传的方式传递至子代中[3]。由于线粒体基因组上的致病性突变可导致多种严重的线粒体代谢相关遗传性疾病[4],因此对全线粒体基因组的高通量测序在线粒体遗传病的诊断和深入研究十分重要。但是,mtDNA突变往往具有异质性[5],且在细胞核基因组中存在同源序列[6],上述特点给全线粒体基因组的特异性深度测序带来了额外的困难。本研究旨在建立了一种基于线粒体分离技术的mtDNA文库构建和测序方法,以期实现特异性针对线粒体基因组的深度测序。
1 材料与方法
1.1主要仪器及试剂 Thermo ST16离心机(美国Thermo公司),Microfuge 20R 4 ℃离心机(德国Beckman公司),ABI Veriti PCR仪(美国ABI公司),CELDOC XR凝胶图像分析仪(美国Bio-Rad公司),Qubit 3.0荧光计(美国Invitrogen公司),A63880 AMPure XP磁珠(德国Beckman公司)。Lymphoprep淋巴细胞分离液、SepMate离心管(加拿大Stemcell公司),NP40裂解液、Tween-20(美国Sigma-Aldrich公司),RSB裂解液[配制:500 μL 10 mmol/L Tris-HCl(pH 7.4),100 μL 10 mmol/L NaCl,150 μL 3 mmol/L MgCl2,混匀后加ddH2O定容至50 mL],2×TD 缓冲液[配制:2 mL 20 mmol/L Tris-HCl(pH 7.6),1 mL 10 mmol/L MgCl2,20 mL mmol/L 20%二甲基甲酰胺(DMF),混匀后加ddH2O定容至100 mL],ExoV核酸外切酶(英国NEB公司),PCR扩增试剂盒、Tn5转座酶试剂盒、双端测序标签(Index)文库制备试剂盒(南京诺唯赞公司),DNA纯化试剂盒(美国Zymo公司)。
1.2样本收集、mtDNA获取及分离纯化 收集2022年于本院志愿体检健康者的外周静脉血样本1 mL,室温保存于EDTA-K2抗凝管中。在8 h内采用SepMate离心管,按照Lymphoprep淋巴细胞分离液说明书分离外周血单个核细胞(PBMC)。设置4个PBMC的裂解条件进行分组:条件1为含5% NP40和1% Tween-20的RSB裂解液;条件2为含5% NP40,不含Tween-20的RSB裂解液;条件3为含1% NP40和1% Tween-20的RSB裂解液;条件4为含1% NP40,不含Tween-20的RSB裂解液。向PBMC中加入15 μL裂解液,冰上裂解3 min,4 ℃、12 000×g离心5 min,立即吸取9.5 μL上清液至新的1.5 mL EP管内。根据ExoV核酸外切酶说明书操作,在上清液中加入1.2 μL缓冲液,1.2 μL ATP和1 μL ExoV,37 ℃反应30 min, 70 ℃反应30 min使ExoV灭活,即得到分离纯化后的mtDNA。
1.3分离纯化后mtDNA的纯度验证 针对核基因组18S rRNA、28S rRNA基因和线粒体基因组m.8363及m.13513位点设计引物,引物序列见表1。PCR反应体系为20 μL,包括分离纯化后的mtDNA 2 μL,2×Taq Master Mix 10 μL,上、下游引物各0.8 μL(引物浓度为10 μmol/L),加入ddH2O补足至20 μL。PCR循环参数:98 ℃预变性30 s;98 ℃变性10 s,63 ℃退火30 s,72 ℃延伸1 min,循环35次;4 ℃保存。PCR反应结束后,对反应产物行20 g/L琼脂糖凝胶电泳,根据是否可见相应片段大小的条带定性判断模板DNA中有无核基因组DNA或线粒体基因组DNA。

表1 引物序列及产物片段大小
1.4建立全线粒体基因组测序文库 在分离纯化后的mtDNA中加入12.4 μL 2×TD 缓冲液,0.5 μL Tn5,70 ℃反应30 min,将mtDNA片段化;使用DNA纯化试剂盒对片段化mtDNA进行纯化,使用双端测序Index文库制备试剂盒进行PCR反应加接头引物,PCR体系为20 μL,包括纯化后的mtDNA 6 μL,上、下游接头引物各2 μL,5×TAB 5 μL,TAE 0.5 μL,加入ddH2O补足至20 μL;PCR循环参数:98 ℃预变性30 s;98 ℃变性10 s,63 ℃退火30 s,72 ℃延伸1 min,循环18次;4 ℃保存。对PCR反应产物进行2轮磁珠回收,取1 μL 回收产物行20 g/L琼脂糖凝胶电泳,若可见185~2 000 bp大小的弥散条带则认为得到合格测序子文库。
1.5高通量测序及数据分析 使用Qubit 3.0荧光计测定子文库的浓度,等量混合得到测序文库。通过Illumina NovaSeq 6000测序系统进行测序,测序策略采用150 bp双端测序法。测序下机数据使用Trim galore、Bowtie2等生物信息学软件完成序列分析和比对,通过GATK标准化线粒体基因组SNP/Indel变异检测流程进行变异位点分析。设置质量控制参数包括:每个样本数据产出量(Clean bases数)>0.4 G,碱基质量值Q30>80,线粒体reads数占比>20%,平均测序深度>5 000×。
1.6Sanger测序 针对样本4 m.10559A>G、m.10398A>G、m.10400C>T位点设计引物,引物序列见表1。 PCR体系为50 μL,包括DNA模板2 μL,2×Taq Master Mix 25 μL,10 μmol/L上、下游引物各2 μL,加入ddH2O补足至50 μL。PCR循环参数:98 ℃预变性30 s;98 ℃变性10 s,63 ℃退火30 s,72 ℃延伸1 min,循环30次;4 ℃保存。PCR产物送上海华大公司测序验证,Sanger测序试剂及仪器由美国 Applied Biosystems 公司提供。
2 结果
2.1mtDNA分离纯化的最优条件及测序结果 对本研究设置的4个分离纯化mtDNA的条件进行验证,电泳结果显示,阳性对照4条泳道全部扩增出,阴性对照4条泳道均未扩增出;条件1~3经PCR 扩增35个循环后,除扩增出线粒体基因组外,仍额外扩增出核基因组18S rRNA、28S rRNA;而条件4仅扩增出线粒体基因组(图1)。

注:琼脂糖凝胶电泳对分离纯化后mtDNA纯度的验证,其中条件1为5% NP40+1% Tween-20;条件2为5% NP40,不含Tween-20;条件3为1% NP40+1% Tween-20;条件4为1% NP40,不含Tween-20;阳性对照模板为全基因组DNA,阴性对照未加模板;泳道1,核基因组18S rRNA;泳道2,核基因组28S rRNA;泳道3,线粒体基因组m.8363位点;泳道4,线粒体基因组m.13513位点。图1 对分离纯化后mtDNA纯度的电泳验证结果
在条件4下,对6例PBMC建库样本进行mtDNA文库构建及深度测序,结果显示6例样本测序结果中,数据产出量最低可达0.45 G,碱基质量值Q30均在85以上,mtDNA reads数占比最高可达92%,平均测序深度最高超过20 000×(表2)。

表2 6例PBMC建库样本测序结果
2.2同质性和异质性变异位点的验证结果 对6例样本的测序结果进行详细分析,每例样本均可检出若干同质性变异(表3、表4),其中样本4检出1个异质性变异(m.10559A>G,突变率为17%),经Sanger测序验证,该样本确实存在相应的异质性变异。同时,针对该样本选取数个同质性变异位点进行Sanger测序验证,结果均可检出相应变异(图2)。

注:A,m.10559A>G为异质性变异,异质度为17%;B,Sanger测序显示m.10559A>G为异质性变异;C,m.8701A>G为同质性变异;D,Sanger测序显示m.8701A>G为同质性变异;E,m.10398A>G为同质性变异;F,Sanger测序显示m.10398A>G为同质性变异;G,10400C>T为同质性变异;H,Sanger测序显示m.10400C>T为同质性变异。图2 Sanger测序验证样本4中的变异位点
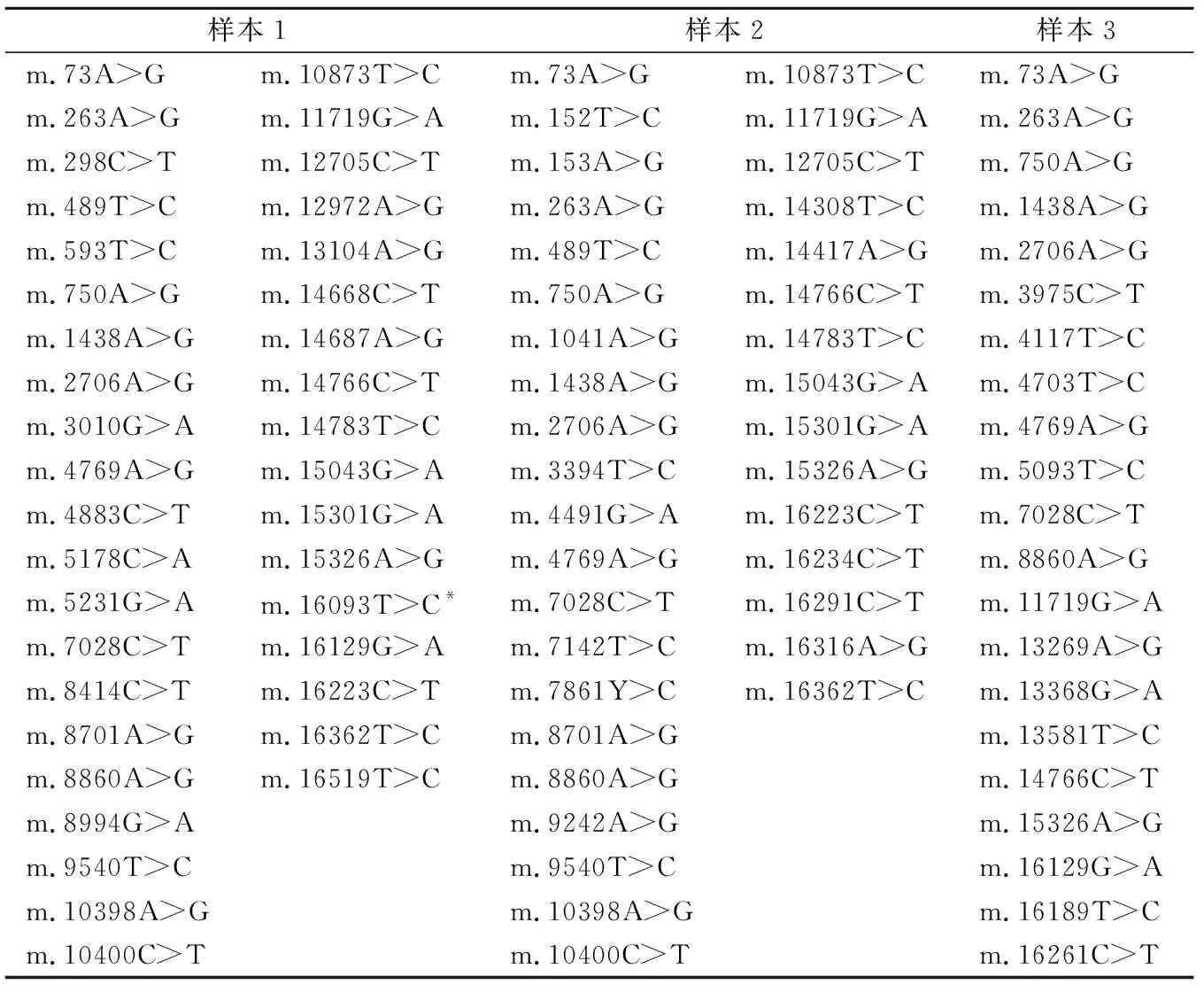
表3 1~3号建库样本所有同质性和异质性变异
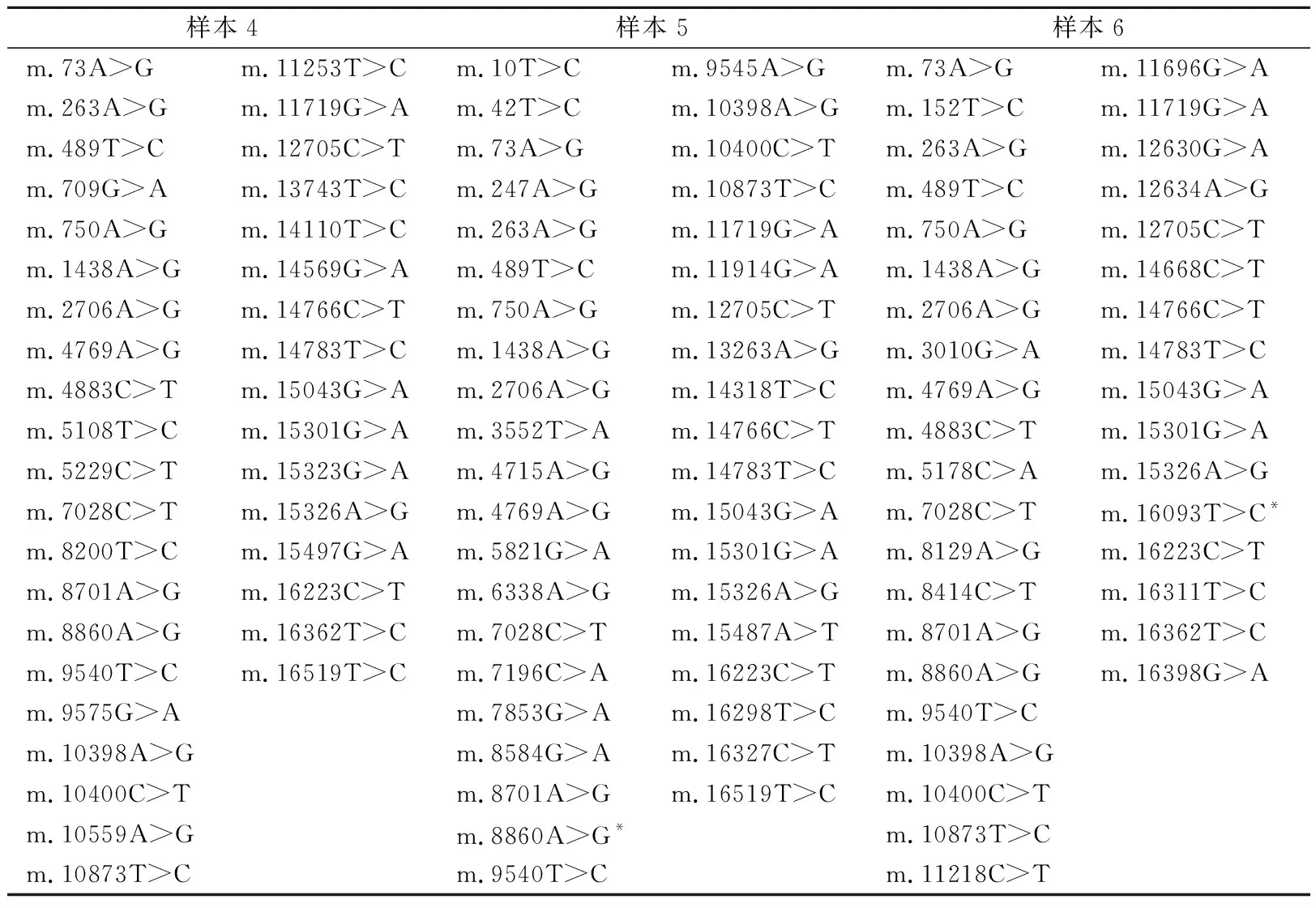
表4 4~6号建库样本所有同质性和异质性变异
3 讨论
本研究创新性地构建了一种基于线粒体分离纯化的全线粒体基因组深度测序方法,并通过核质分离的方式来实现非PCR富集mtDNA。通过扩增核基因组18S rRNA、28S rRNA以及线粒体基因组m.8363和m.13513位点检测mtDNA分离后的纯度,证明本方法可获得较高纯度的mtDNA,有效避免了文库中出现细胞核DNA及“核线粒体同源序列”的干扰;通过Sanger测序,笔者进一步验证了本方法所检出的同质性变异和异质性变异的准确性,表明构建的深度测序方法结果准确可信。然而,本方法仍存在一定局限性:在核质分离时需要保证细胞核的完整,因此,检测时需要使用采血8 h内的新鲜外周血,而不可使用冻存的血细胞样本。此外,在受检样本类型为非外周血细胞时(如尿液细胞、肌肉组织、肿瘤组织等),本方法的有效性尚不明确,需在后续工作中展开进一步研究,以获得相应结论。
线粒体是真核生物细胞中最重要的细胞器之一,其携带的线粒体基因组具有多拷贝、易突变等特征,对线粒体发挥正常生理功能至关重要。因此,获得高质量的mtDNA序列信息在临床和科研中具有重要价值。传统的Sanger测序方法是针对特定位点检测的金标准[7],但受测序长度限制,Sanger测序很难应用于全线粒体基因组测序,且无法分辨低异质度的变异位点。随着高通量测序技术[8]的出现,对全线粒体基因组进行深度测序已成为获得mtDNA序列信息的最优方法。目前,主流的全线粒体基因组高通量测序策略包括全基因组测序策略和mtDNA富集策略。全基因组测序策略通过筛选、拼装全基因组测序[9]或外显子测序[10]的数据中比对线粒体基因组上的reads,获得全线粒体基因组的序列信息,但是该策略成本较高,难以大规模应用,且测序结果难以排除核基因组同源序列的干扰。mtDNA富集策略主要包括长片段PCR富集法和特异性探针捕获法。长片段PCR富集法[11]通过两对或多对引物扩增全线粒体基因组,再将扩增得到的长片段mtDNA打断进行深度测序,这种方法可以对全线粒体基因组进行深度测序,但是同样受到潜在的核基因组同源序列干扰。此外,长片段PCR经常存在扩增失败的情况,而PCR本身存在的偏好性也会对异质性变异的测序结果造成影响。特异性探针捕获法[12]需要设计大量与mtDNA互补的寡核苷酸探针,通过PCR加接头引物来富集mtDNA片段进行深度测序,此方法相对复杂,且同样无法避免核基因组同源序列的干扰。相比于以上方法的优缺点,本研究创新性地构建了一种基于线粒体分离纯化技术的全线粒体基因组深度测序的方法,通过核质分离,可有效获得mtDNA特异性文库。本方法整个实验流程通过非PCR的方式来富集mtDNA,避免了PCR的偏好性对测序结果的影响,且可以避免核基因组同源序列的干扰。在具体实验操作上,本方法大大简化了实验流程,较其他mtDNA高通量测序方案耗时短,reads利用率高,建库成本较低,可以应用于大规模人群mtDNA的测序工作。
目前线粒体疾病(mitochondrial diseases,MD)的诊断仍具有很大的挑战性。虽然一些MD具有明确的临床症状,容易识别,如慢性进行性眼外肌麻痹(chronic progressive external ophthalmoplegia,CPEO)[13]、线粒体脑肌病合并乳酸酸中毒和中风样发作(mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes,MELAS)[14]等,但更多的患者不会表现出疾病的典型症状和体征。MD的实验室诊断多数是通过对疾病受累的组织或器官进行生物化学(如测定线粒体ATP酶的活性)等方法来诊断,最终确诊常需要侵入性活检,耗时长且不易被患者接受。迄今为止,研究者已发现了300多个基因的致病性突变可导致MD[15],Mitomap数据库也给出96个明确致病的mtDNA突变位点。当患者临床症状为多系统受累或综合征表现时,需要高度怀疑MD,则应优先进行基因检测[16]。对于MD的遗传学病因诊断,本方法由于建库实验流程简易、成本低、准确性高,相较于其他测序方法具有较大优势,可用于研究MD患者的致病机制,发现潜在性MD患者,同时也可以指导临床医生给予患者精确的遗传咨询、产前诊断等方面的帮助。
综上所述,本研究开发了一种基于线粒体分离纯化技术的mtDNA建库及测序方法,可实现特异性全线粒体基因组深度测序。本研究通过非PCR的方法获得较纯的mtDNA文库,不易受核基因组同源序列干扰,可准确且灵敏地检出线粒体基因组的同质性和异质性变异。此外,本方法可较好地应用于PBMC样本,适合在临床上推广和应用。
猜你喜欢
海洋通报(2021年1期)2021-07-23 01:55:14
生物学通报(2021年4期)2021-03-16 05:41:26
趣味(数学)(2020年4期)2020-07-27 01:44:16
支部建设(2020年15期)2020-07-08 12:34:32
职教论坛(2017年4期)2017-03-13 16:43:19
百科知识(2015年18期)2015-09-10 07:22:44
财经科学(2014年7期)2015-04-20 20:48:44
职业技术教育(2014年7期)2014-08-15 19:35:45
癌变·畸变·突变(2014年1期)2014-03-01 04:39:36
植物营养与肥料学报(2011年2期)2011-10-26 03:52:48
