
Solriamfetol impurities: Synthesis, characterization, and analytical method (UPLC-UV) validation
2023-05-29 10:00NafisahAlRifaiAnasAlshishaniFouaDarrasOlaTahaShereenAuJallouLenaShaghlilYousefAlEini
Nafisah Al-Rifai , Anas Alshishani , Foua Darras , Ola Taha , Shereen Au-Jallou ,Lena Shaghlil , Yousef Al-Eini
a Pharmaceutical and Chemical Engineering Department, School of Medical Sciences, German Jordanian University, Amman, Jordan
b Faculty of Pharmacy, Zarqa University, Zarqa, Jordan
c Research and Development Department, Resonance Research Lab, Amman, Jordan
d Department of Pharmaceutics, College of Pharmacy, King Khalid University, Abha, Saudi Arabia
Keywords:
Solriamfetol
Impurity analysis
Impurity synthesis
UPLC
Method validation
ABSTRACT Given that impurities may affect the quality and safety of drug products, impurity identification and profiling is an integral part of drug quality control and is particularly important for newly developed medications such as solriamfetol,which is used to treat excessive daytime sleepiness.Although the highperformance liquid chromatography analysis of commercial solriamfetol has revealed the presence of several impurities, their synthesis, structure elucidation, and chromatographic determination have not been reported yet.To bridge this gap, we herein identified, synthesized, and isolated eight processrelated solriamfetol impurities, characterized them using spectroscopic and chromatographic techniques,and proposed plausible mechanisms of their formation.Moreover,we developed and validated a prompt impurity analysis method based on ultrahigh-performance liquid chromatography with UV detection, revealing that its selectivity, linearity, accuracy, precision, and quantitation limit meet the acceptance criteria of method validation stipulated by the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use.Thus,the developed method was concluded to be suitable for the routine analysis of solriamfetol substances.
1.Introduction
Solriamfetol hydrochloride (R-2-amino-3-phenylpropylcarbamate hydrochloride, Fig.1) is a central nervous system drug that is used to treat excessive daytime sleepiness accompanied by narcolepsy or obstructive sleep apnea and has been marketed since late 2019 by Jazz Pharmaceuticals under the brand name Sunosi [1,2].The action mechanism of solriamfetol is not yet fully understood and is thought to involve the inhibition of dopamine and norepinephrine reuptake[3-5].

Fig.1 .Structure of solriamfetol hydrochloride.
Fig.2 A presents the method used to synthesize solriamfetol as an active pharmaceutical ingredient(API)according to a patent filed by SK Biopharmaceuticals [6].This method involves the protection of the amino group of D-phenylalaninol followed by the introduction of a carbamate moiety at the hydroxyl group using phosgene and ammonia, with subsequent hydrogenation-induced deprotection and salt formation affording solriamfetol in the form of a hydrochloride.An alternative one-step synthesis of solriamfetol in high yield,which involves reacting D-phenylalaninol with sodium cyanate under acidic conditions[7],is described in Fig.2B.
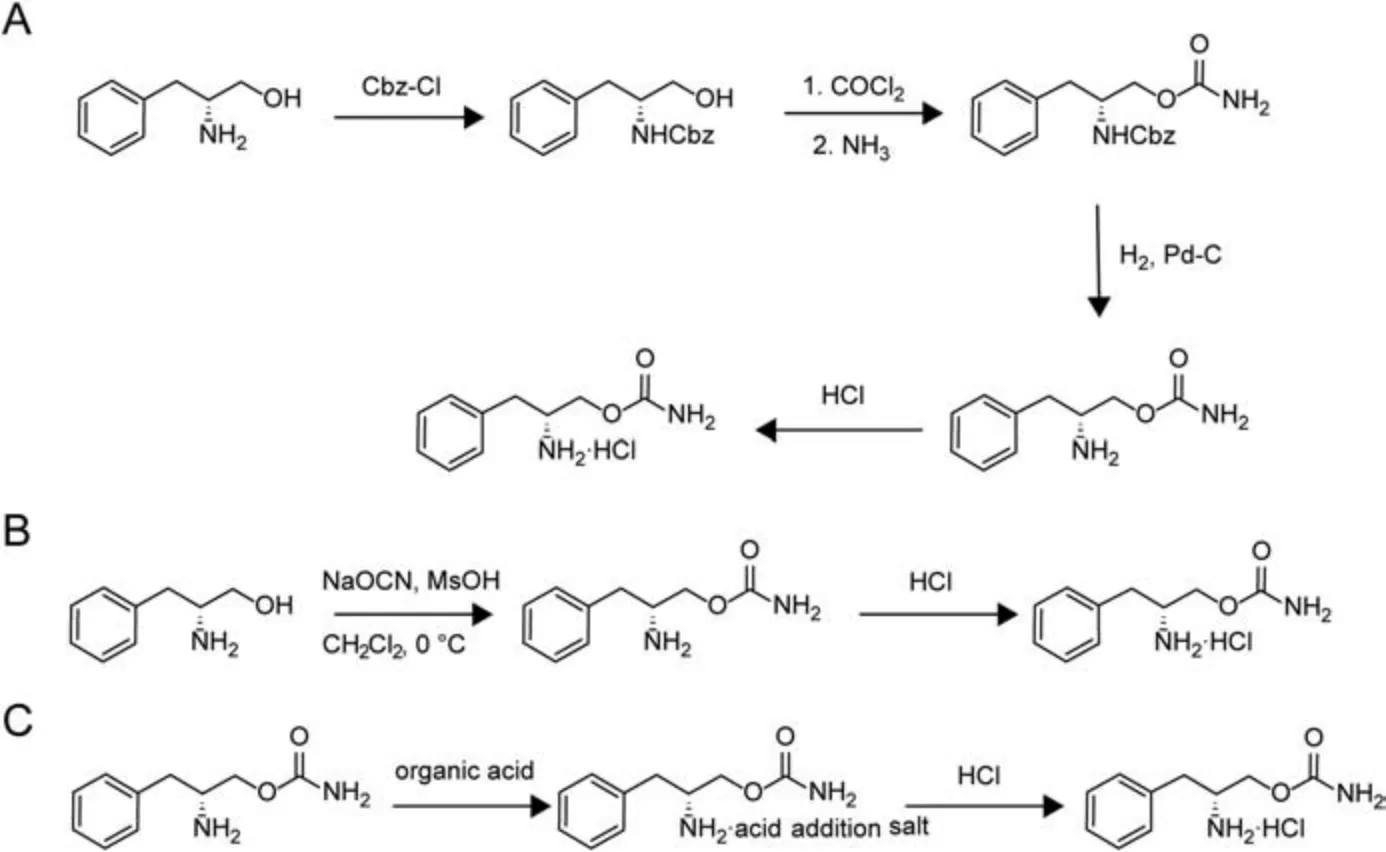
Fig.2 .Previously reported solriamfetol synthesis methods.(A) Patent US5955499 A [6], (B) patent WO2005033064 A1 [7], and (C) patent WO2020035769 A1 [8].Cbz: benzyloxycarbonyl; Ms: mesyl.
Patent WO2020035769A1 describes an improved high-yield synthesis of high-purity solriamfetol hydrochloride without forming any isomers and other process-related impurities [8].In this patent,it is reported that solriamfetol free base can be reacted with an organic acid and then converted to the hydrochloride salt(Fig.2C).
However, process-related impurities are commonly detected by high-performance liquid chromatography (HPLC) during the synthesis and purification steps.As these impurities may affect the quality and safety of drug products, impurity identification and profiling have received considerable attention from regulatory authorities [9].The International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use(ICH)states that impurities present in quantities above the identification thresholdshould beidentified andcharacterized[10].AllAPIs usedin human medication must meet the ICH qualityguidelines.The quality of any API depends on its synthetic process, potential degradation pathway, and possible side reactions.Consequently, API manufacturers attempt to minimize impurity levels; nevertheless, the formation of impurities cannot be fully avoided.There have been many reports on the identification and characterization of unknown impurities formed in drug development processes [11-15].If some of these characterized impurities are not readily available,appropriate synthetic procedures should be established to produce quantities sufficientforthedevelopmentandvalidation of ananalyticalmethod and thus benefit pharmaceutical development teams worldwide.
The marketed solriamfetol drug is the R-enantiomer, while the S-enantiomer might exist as a chiral impurity [16].Phenylalaninol enantiomers are also considered potential impurities of solriamfetol and can be present either as the residual starting material of the synthesis and/or as degradation products of solriamfetol[17].Patent WO2020035769A1 reports nine process-related impurities of solriamfetol potentially produced during its synthesis [8](Fig.S1).Moreover, patent WO2021250067A2 describing solriamfetol purification reports five most common and critical solriamfetol impurities (Fig.S1) and suggests mechanisms of their formation [18].However, these patents do not deal with the syntheses,structure elucidation,or chromatographic determination of solriamfetol impurities.
A dispersion of X (1.0 g, 2.1 mmol) in methanol (15.0 mL) was treated with 10% (m/m) Pd/C (200 mg) and stirred at 60-70°C under H2for 2 h.Subsequently, heating was stopped, and the reaction mixture was stirred for another 12 h and filtered.The filtrate was evaporated under reduced pressure to give an oily material(0.620 g,79.5% yield)that was dissolved in methanol.The solution was dropwise supplemented with aqueous hydrochloric acid(0.5 mL)until a precipitate was formed.The precipitate was filtered off and dried to afford imp.5 (0.286 g, 36% yield, 95.3% purity by HPLC) as an off-white solid.1H NMR (400 MHz, DMSO-d6): δ/ppm = 8.82 (d, J = 8.3 Hz,1H), 8.22 (s, 3H), 7.29-7.20 (m, 9H, Ar),7.06-7.04 (m, 2H), 6.55 (bs, 2H), 4.14-4.13 (m,1H), 3.98 (bs,1H),3.93-3.81(m,2H),2.93(dd,J=13.9,5.3 Hz,1H),2.81-2.72(m,2H),2.62 (dd, J = 13.5, 9.1 Hz, 1H);13C NMR (101 MHz, DMSO-d6):δ/ppm = 167.7 (NC=O),156.5 (OC=O),138.1 (C, Ar),134.8 (C, Ar),129.5 (2CH, Ar),129.2 (2CH, Ar),128.4 (2CH, Ar),128.3 (2CH, Ar),127.0(CH,Ar),126.3(CH,Ar),64.4(CH2O),53.3(CHN),50.0(CHN),36.7 (PhCH2), 36.5 (PhCH2); HRMS (ESI+): calcd for C19H24N3O3+:342.18120 [M+H]+; found 342.1808.
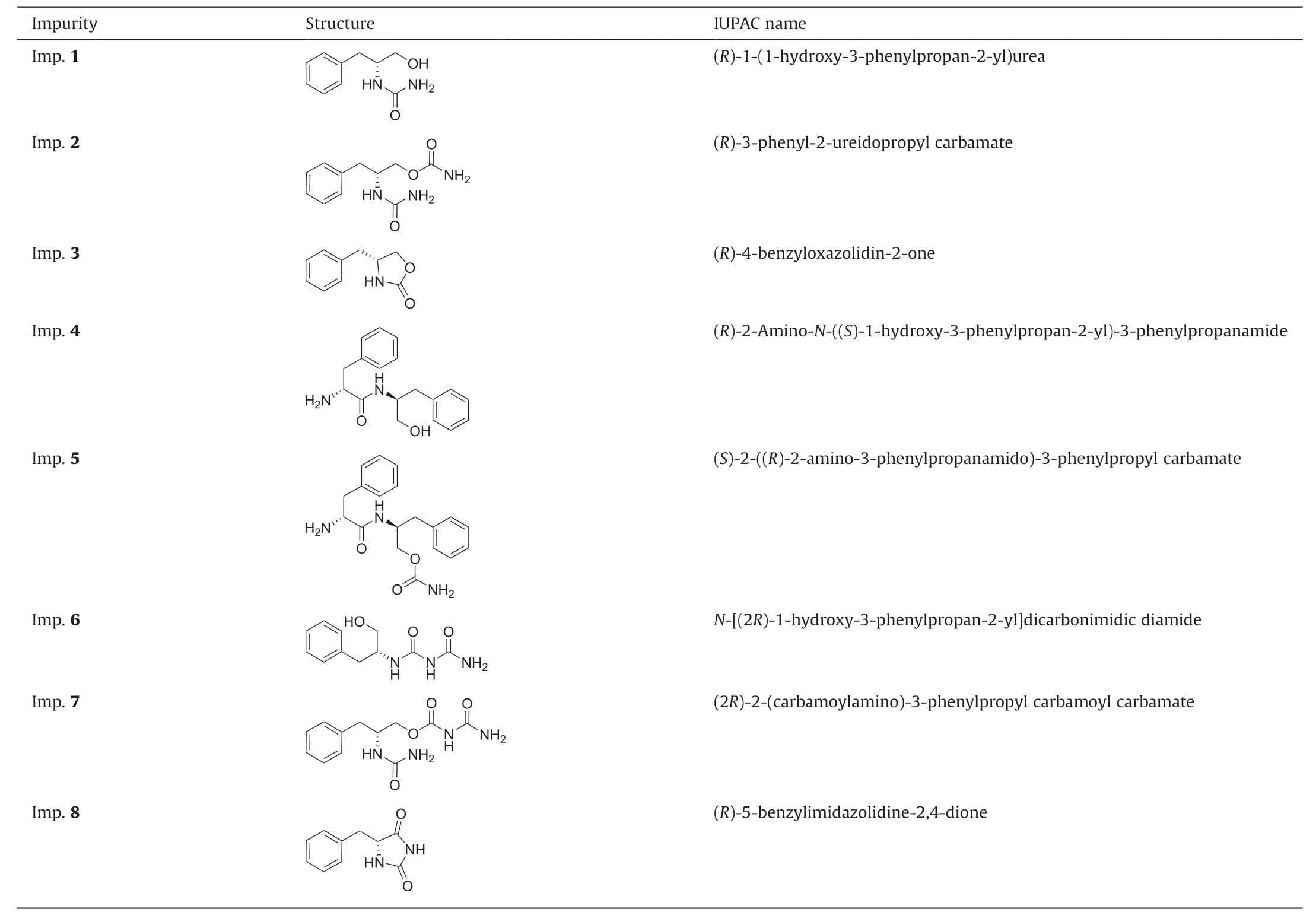
Table 1 International Union of Pure and Applied Chemistry (IUPAC) names and structures of solriamfetol impurities investigated hereina.
2.Experimental
2.1.Materials and reagents
D-phenylalaninol (I, 98%), N-Cbz-D-phenylalanine (Z-D-Phe-OH)(II, 97%), L-phenylalaninol (VII, 97%), and biuret (XI, 98%) were purchased from AA Blocks(San Diego,CA,USA).Other chemicals and reagents were acquired from commercial sources including Merck(Darmstadt,Germany),Quality Reagent Chemicals(QReC;Auckland,New Zealand),and TEDIA(Fairfield,OH,USA).Silica gel(Geduran Si 60; 0.063-0.200 mm) from Merck was used for column chromatography.Ultrapure water (18.2 MΩ cm) was generated using a Millipore water purification system(Molsheim,France)and used to prepare mobile phases for HPLC.
由图2b可知:3410.11 cm-1νOH、2929.84 cm-1νC-H、1429.23 cm-1δC-H、1732.05 cm-1νC=O、1068.55 cm-1νC-O和1010.68 cm-1νC-O的吸收,表明72%vol红枣白兰地致浊物中可能含有糖类化合物。3410.11 cm-1νOH、1642 cm-1νC=C、1271.07 cm-1ν=C-O,表明 72%vol酒的致浊物中可能含有水溶性的酚类化合物。结果表明,高度酒的致浊物可能是糖类及水溶性酚类化合物。
Selectivity was evaluated by injecting a blank solution and a 1 mg/mL solution of solriamfetol spiked with eight impurities (1 μg/mL each)and was verified by the absence of interference between blank peaks and analyte peaks as well as by the proper separation between the peaks of solriamfetol and those of the eight impurities (Fig.4).The corresponding resolutions ranged from 2.3 to 10.(Table 2).
2.2.HPLC
Chromatographic separation was achieved using an HPLC instrument consisting of an LC-40D XR pump,SIL-40C XR autosampler,CTO-40 S column oven, and SPD-M30A PDA detector and equipped with an 8-cm-path-length flow cell (Shimadzu, Kyoto, Japan).The detection wavelength was set to 210 nm.Separation was achieved at 30°C and a flow rate of 0.4 mL/min using a Kinetex Polar C18column(100 mm 2.1 mm, 2.6 μm) manufactured by Phenomenex (Torrance, CA, USA).Elution was performed in gradient mode using mobile phases A(0.1% aqueous perchloric acid)and B(0.1 M aqueous perchloric acid:acetonitrile,10:90, V/V).Mobile phase B was maintained at 3% for 13 min, changed to 20% from 13 to 16 min, and maintained at 20% for 6 min.The column was re-equilibrated at the initial ratio for 7 min.The injection volume equaled 1 μL.Data were processed using Lab Solution software version 6.106SP1(Shimadzu).
2.3.Liquid chromatography-mass spectrometry (LC-MS)
MS/MS identification was conducted using a quadrupole timeof-flight mass spectrometer (Triple TOF 5600; AB Sciex, Foster City, CA, USA) equipped with an electrospray ionization (ESI)source.Scanning was performed within the m/z range of 100-1500.The pressures of ion source gases one and two were set to 379 kPa,and the curtain gas pressure was set to 241 kPa.The collision energy was set to 35 eV.Nitrogen was used as the collision cell,nebulizer and auxiliary gas.Data were acquired using the Analyst®TF 1.6 software (AB Sciex).
2.4.Nuclear magnetic resonance (NMR) spectroscopy
NMR spectra were recorded on a 400-MHz FT-NMR spectrometer (Avance-III, Bruker, Germany) using deuterated dimethyl sulfoxide (DMSO-d6) as the solvent and tetramethylsilane as the internal standard.
2.5.Fourier transform infrared (FTIR) spectroscopy
2.6.3.2.Preparation of benzyl (R)-(1-hydroxy-3-phenylpropan-2-yl)carbamate (IV).III(3.550 g,11.3 mmol)was dissolved in methanol(40 mL) inside a 250 mL RBF, and the solution was portionwise supplemented with sodium borohydride(2.95 g,77.9 mmol)upon stirring in an ice bath.The mixture was further stirred for 12 h at room temperature, concentrated under reduced pressure, and the residue was treated with water and extracted with ethyl acetate.The combined organic extracts were dried over anhydrous sodium sulfate and concentrated under reduced pressure to obtain IV(2.495 g, 78% yield) as a white solid.
2.6.Synthesis and characterization of solriamfetol impurities
Imps.1-8 were synthesized as described in Fig.3.
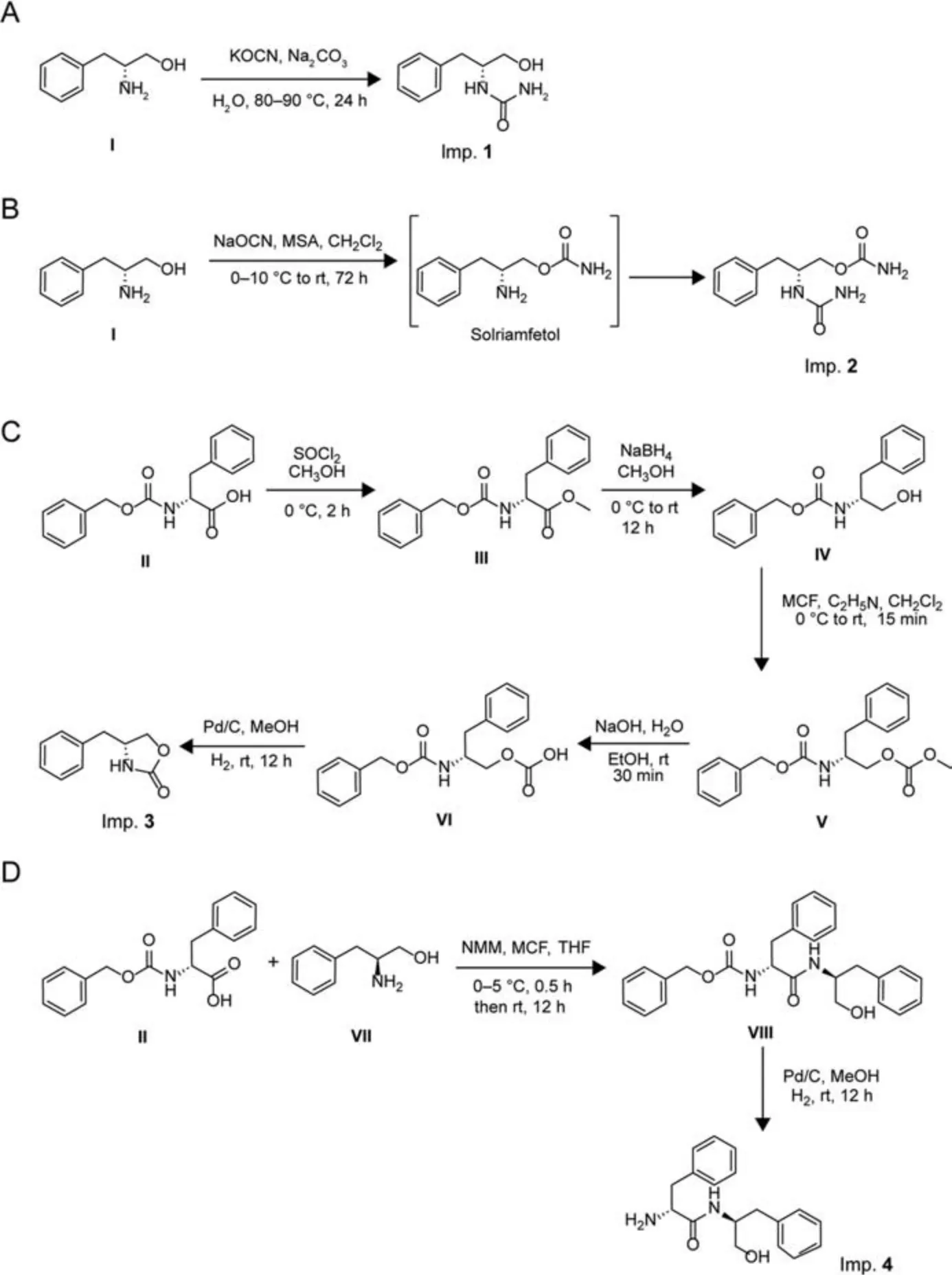
Fig.3 .Syntheses of imps.1-8.(A) Imp.1 ((R)-1-(1-hydroxy-3-phenylpropan-2-yl)urea) from compound I, (B) Imp.2 ((R)-3-phenyl-2-ureidopropyl carbamate) from compound I,(C) Imp.3((R)-4-benzyloxazolidin-2-one) in five steps starting with compound II, (D) Imp.4 ((S)-2-amino-N-((R)-1-hydroxy-3-phenylpropan-2-yl)-3-phenylpropanamide) in two steps starting with compounds II and VII,(E)Imp.5((R)-2-((R)-2-amino-3-phenylpropanamido)-3-phenylpropyl carbamate)in three steps starting with compound VII,(F) Imp.6(N-[(2R)-1-hydroxy-3-phenylpropan-2-yl]dicarbonimidic diamide) in two steps starting with compound XI, (G) Imp.7 ((2R)-2-(carbamoylamino)-3-phenylpropyl carbamoyl carbamate) from imp.2, and (H) Imp.8 ((R)-5-benzylimidazolidine-2,4-dione) from compound XIII.MSA: methanesulfonic acid; MCF: methyl chloroformate; NMM: Nmethylmorpholine.

Fig.3 .(continued).
2.6.1.(R)-1-(1-hydroxy-3-phenylpropan-2-yl)urea (imp.1)
I (0.500 g, 3.31 mmol) was dissolved in H2O (20.0 mL) inside a 250 mL round-bottom flask(RBF),which was subsequently charged with a solution of sodium carbonate (0.250 g, 2.36 mmol) in H2O(2.5 mL)and potassium cyanate(0.500 g,6.16 mmol).The reaction mixture was stirred overnight at 80-90°C and monitored using HPLC.After the reaction was complete, the mixture was extracted with methylene chloride, and the combined organic extracts were dried over anhydrous sodium sulfate and concentrated under reduced pressure to obtain imp.1(0.366 g,57% yield,99.3% purity by HPLC) as a white solid.1H NMR (400 MHz, DMSO-d6): δ/ppm = 7.28-7.17 (m, 5H, Ar), 5.85 (d, J = 7.6 Hz, 1H), 5.42 (d,J = 7.1 Hz, 2H), 4.81-4.77 (m,1H), 3.70 (s,1H), 3.29 (dt, J = 28.6,5.6 Hz, 2H), 2.81-2.50 (m, 2H);13C NMR (101 MHz, DMSO-d6): δ/ppm=158.4(C=O),139.4(C,Ar),129.2(2CH,Ar),128.1(2CH,Ar),125.8 (CH, Ar), 62.7 (CH2OH), 52.5 (CHNH), 37.3 (PhCH2); HRMS(ESI+):calcd for C10H15N2O2+:195.11280[M+H]+;found 195.1123.
2.6.2.(R)-3-Phenyl-2-ureidopropyl carbamate (imp.2)
I (16.0 g, 105.8 mmol) was dissolved in methylene chloride(150 mL)inside a 250 mL RBF,and the solution was supplemented with sodium cyanate (17.0 g, 2611.49 mmol), cooled to 0°C, and dropwise supplemented with methanesulfonic acid(23.68 g,16 mL,246.4 mmol)at 0°C.After the addition was complete,the mixture was stirred overnight at room temperature, and the reaction was monitored by HPLC.When the reaction was complete, the solvent was concentrated under reduced pressure, the residue was dissolved in H2O(~70 mL),and the mixture was sonicated for 10 min.The produced crude white solid was collected by filtration and dried (13.0 g, 52% yield).For purification, the crude product was refluxed in ethyl acetate:methanol(1:1,V/V,30 mL)for 1 h,filtered,and dried to yield imp.2 as a white solid(98.3% purity by HPLC).1H NMR(400 MHz,DMSO-d6):δ/ppm=7.31-7.19(m,5H,Ar),6.53(bs,2H), 5.92 (d, J = 8.2 Hz, 1H), 5.45 (s, 2H), 3.94-3.88 (m, 1H),3.83-3.74 (m, 2H), 2.78-2.65 (m, 2H);13C NMR (101 MHz,DMSO-d6): δ/ppm = 158.0 (C=O), 156.7 (C=O), 138.5 (C, Ar),129.1(2CH, Ar), 128.2 (2CH, Ar), 126.1 (CH, Ar), 64.9 (CH2O), 49.8(CHNH), 37.45 (PhCH2); HRMS (ESI+): calcd for C11H16N3O3+:238.11662 [M+H]+; found 238.1189.
2.6.3.(R)-4-Benzyloxazolidin-2-one (imp.3)
VI (1.18 g, 3.6 mmol) was dissolved in methanol (20.0 mL),10%(m/m) Pd/C (120 mg) was added, and the mixture was stirred at room temperature under H2for 12 h.The mixture was filtered,and the filtrate was evaporated under reduced pressure.The residue was crushed with a spatula to give imp.3(0.367 g,52% yield,100% purity by HPLC) as a white solid.1H NMR (400 MHz, DMSO-d6):δ/ppm = 7.79 (s,1H), 7.32-7.21 (m, 5H, Ar), 4.25 (t, J = 8.2 Hz,1H),4.08-4.02(m,1H),3.98(dd,J=8.2,5.4 Hz,1H),2.84-2.72(m,2H);13C NMR(101 MHz,DMSO-d6):δ/ppm=158.6(C=O),136.6(C,Ar),129.4 (2CH, Ar),128.4 (2CH, Ar),126.5 (CH, Ar), 68.0 (CH2O), 52.5(CHNH), 40.25 (PhCH2); HRMS (ESI+): calcd for C10H12NO2+:178.08626 [M+H]+; found 178.0857.
2.6.3.1.Preparation of methyl ((benzyloxy)carbonyl)-d-phenylalaninate (III).II (3.0 g, 10.0 mmol) was dissolved in methanol(50.0 mL, 0.2 M) inside a 250 mL RBF.The solution was dropwise supplemented with thionyl chloride (1.7 g,14.0 mmol) over 5 min at 0°C upon stirring and further stirred for ~2 h at the same temperature.The reaction mixture was concentrated under reduced pressure, and the residue was treated with water and extracted with methylene chloride.The combined organic extracts were dried over anhydrous sodium sulfate and concentrated under reduced pressure to obtain III(3.550 g,113% yield)as a colorless oil.
The spectra of neat samples were recorded in attenuated total reflectance mode on an FTIR-4X spectrometer (Jasco, Hachioji,Tokyo,Japan).
2.6.3.3.Preparation of benzyl (R)-(1-((methoxycarbonyl)oxy)-3-phenylpropan-2-yl)carbamate (V).IV (1.5 g, 5.3 mmol) was dissolved in methylene chloride (40.0 mL) inside a 250 mL RBF, and the solution was supplemented with pyridine(1.5 mL,18.6 mmol).Subsequently, methyl chloroformate (2.43 mL, 31.5 mmol) was dropwise added upon cooling in an ice bath, and the reaction mixture was stirred for 15 min at room temperature and washed with water.The methylene chloride layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure to obtain crude V (1.5 g, 83% yield).
2.6.3.4.Preparation of (R)-2-(((benzyloxy)carbonyl)amino)-3-phenylpropyl hydrogen carbonate (VI).Crude V (1.5 g, 4.4 mmol)was dissolved in ethanol (20.0 mL) inside a 250 mL RBF, and the dispersion was charged with a solution of sodium hydroxide (1.3 g,32.5 mmol)in water(10.0 mL)and ethanol(10.0 mL)upon stirring.After ~30 min stirring at room temperature, ethanol was removed under reduced pressure, water was added, and pH was adjusted to 3.0 with aqueous hydrochloric acid.The mixture was then extracted with methylene chloride, and the combined organic extracts were dried over anhydrous sodium sulfate and concentrated under reduced pressure.The resulting product was purified by column chromatography using methylene chloride:methanol (50:1, V/V) as an eluent to afford VI(1.18 g,81% yield)as a white solid.
2.6.4.(R)-2-amino-N-((S)-1-hydroxy-3-phenylpropan-2-yl)-3-phenylpropanamide (imp.4)
A dispersion of VIII(1.0 g,2.3 mmol)in methanol(15.0 mL)was supplemented with 10% (m/m) Pd/C (250 mg), stirred at room temperature under H2for 12 h, and filtered through a filter paper.The filtrate was evaporated under reduced pressure to give imp.4(0.77 g, 81% yield, 99.4% purity by HPLC) as a white solid.1H NMR(400 MHz, DMSO-d6): δ/ppm = 7.77 (d, J = 8.3 Hz,1H), 7.28-7.13(m, 10H, Ar), 4.81 (t, J = 5.6 Hz, 1H), 3.91 (d, J = 5.6 Hz, 1H),3.38-3.26 (m, 3H), 2.85-2.79 (m, 2H), 2.51-2.45 (m, 2H),1.58 (s,2H);13C NMR(101 MHz,DMSO-d6):δ/ppm=174.3(C=O),139.5(C,Ar),139.2 (C, Ar),129.8 (2CH, Ar),129.6 (2CH, Ar),128.6 (2CH, Ar),128.5 (2CH, Ar),126.5 (CH, Ar),126.4 (CH, Ar), 62.8 (CH2OH), 56.5(CHN),52.5(CHN),41.6(PhCH2),37.1(PhCH2);HRMS(ESI+):calcd for C18H23N2O2+: 299.17540 [M+H]+; found 299.1730.
2.6.4.1.Preparation of benzyl ((R)-1-(((S)-1-hydroxy-3-phenylpropan-2-yl)amino)-1-oxo-3-phenylpropan-2-yl)carbamate(VIII).II (3.0 g, 10.0 mmol) was dissolved in tetrahydrofuran(25.0 mL)inside a 250 mL RBF,and the solution was supplemented with N-methylmorpholine (1.1 mL, 10.0 mmol) and methyl chloroformate(770 μL,10.0 mmol)upon stirring.The mixture was further stirred at 0-5°C for 0.5 h, treated with VII (1.8 g,12.0 mmol), and further stirred for 12 h at room temperature.Subsequently, tetrahydrofuran was removed under reduced pressure, and the crude product was taken up in methylene chloride.The solution was washed with 4 M hydrochloric acid and brine,and the organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure to give VIII(4.27 g,99% yield).
2.6.5.(S)-2-((R)-2-amino-3-phenylpropanamido)-3-phenylpropyl carbamate hydrochloride (imp.5)
Herein, we present a first-time account of the syntheses,identification, and characterization of eight potential process-related solriamfetol impurities (Table 1) [19-21] and discuss their formation mechanisms.Moreover,we describe the development and validation of a chromatographic method for analyzing solriamfetol impurities to facilitate their detection and quantitation in industrial settings.
二是虚假信息的泛滥。网络上充斥着大量虚假新闻、虚假事件以及损害他人利益、损害国家名誉等的不良信息,影响公众对舆论的判断力。一些违法分子甚至在网络上发布凭空捏造一些子虚乌有的事件,煽动公众情绪,尤其是煽动公众损害他人利益的信息、影响政府声誉的信息、甚至是危害国家安全的信息,对社会公共安全形成威胁。
2.6.5.1.Preparation of (S)-2-amino-3-phenylpropyl carbamate (IX).VII(1.0 g,6.6 mmol)was dissolved in methylene chloride(25.0 mL)inside a 100 mL RBF, and the solution was sequentially supplemented with sodium cyanate (1.0 g, 15.4 mmol) and methanesulfonic acid (1.5 mL, 23.1 mmol; dropwise over 15 min at 0-5°C).The mixture was stirred for 2 h at the same temperature and concentrated under reduced pressure.The residue was treated with water, and the mixture was pH-adjusted to 10.0 with 1 N sodium hydroxide and extracted with methylene chloride.The combined organic extracts were dried over anhydrous sodium sulfate and concentrated under reduced pressure to obtain an oily material (1.2 g, 93.8% yield).The crude product was purified by columnchromatographyusingmethylenechloride:methanol:ammonia (20:1:0.1, V/V/V) as an eluent to afford IX(0.800 g, 62.5% yield) as a yellow oil.
2.6.5.2.Preparation of benzyl ((R)-1-(((R)-1-(carbamoyloxy)-3-phenylpropan-2-yl)amino)-1-oxo-3-phenylpropan-2-yl)carbamate(X).II(1.7 g,5.7 mmol)was dissolved in tetrahydrofuran(20.0 mL)inside a 250 mL RBF, and the solution was sequentially supplemented with N-methylmorpholine(960 μL,8.7 mmol) and methyl chloroformate(410 μL,5.3 mmol)upon stirring and further stirred at 0-5°C for 0.5 h.Subsequently, IX (0.8 g, 4.1 mmol) was added,and the reaction mixture was stirred for 12 h at room temperature and then concentrated under reduced pressure.The residue was taken up in methylene chloride,and the solution was washed with 4 M hydrochloric acid and brine, dried over anhydrous sodium sulfate, and concentrated under reduced pressure to afford an offwhite solid (4.4 g).The crude product was purified by column chromatography using methylene chloride:methanol:ammonia(30:1:0.1, V/V/V) as the eluent to obtain X (2.1 g, 77.8% yield) as a white solid.
2.6.6.N-[(2 R)-1-Hydroxy-3-phenylpropan-2-yl]dicarbonimidic diamide (imp.6)
I(1.0 g,6.6 mmol)and XII(2.0 g,13.5 mmol)were added to water(~20 mL) inside a 100 mL RBF, and the mixture was stirred at 80-90°C for 72 h and then extracted with methylene chloride and ethyl acetate.The combined organic extracts were dried over anhydrous sodium sulfate and concentrated under reduced pressure to give an oily material (1.3 g, 83% yield), which was subjected to column chromatography using an ethyl acetate:n-hexane gradient of 1:1 to 5:1(V/V)to afford imp.6(0.462 g,29% yield,95.0% purity by HPLC)as a white solid.1H NMR(400 MHz,DMSO-d6):δ/ppm=8.48(s, 1H), 7.55 (bs, 1H), 7.30-7.17 (m, 5H, Ar), 6.69 (s, 2H), 4.89 (t,J=5.2 Hz,1H),3.82(d,J=6.8,1H),3.35-3.33(m,2H),2.85-2.63(m,2H);13C NMR (101 MHz, DMSO-d6): δ/ppm = 155.4 (C=O), 154.0(C=O),138.8(C,Ar),129.2(2CH,Ar),128.2(2CH,Ar),126.1(CH,Ar),61.9 (CH2O), 52.4 (NCH), 37.0 (PhCH2); HRMS (ESI+): calcd for C11H16N3O3+:238.11862[M+H]+;found 238.1185.
2.6.6.1.Preparation of nitrobiuret (XII).XI (5.0 g, 48.5 mmol) was portionwise added to a mixture of concentrated sulfuric (12.5 mL,233.2 mmol) and nitric (3.3 mL, 73.9 mmol) acids at 5 to 0°C inside a 250 mL RBF, and the mixture was stirred under N2at the same temperature for 12 h, poured into ice water, and stirred for 5 min.The precipitate was filtered off and dried to give XII (6.4 g,89% yield) as a white solid.
基于大数据理念的调查研究,不仅可以在就某一确定的主题开展问卷调查的宏观框架下保持一定的导向性,而且具有一定的开放性。这种研究还有利于全面了解调查对象的生成性资源,发掘被调查问卷的设计者忽略的因素。特别是通过学校教师专业发展评价系统和教学信息评价系统获得的调查数据,如“极课大数据”平台等多个渠道提供的数据信息,不仅能体现出信息来源的多维性,而且有利于修正教师专业发展评价要素,以及调整评价系统中子模块的权重。
3.4.4.Sensitivity (LOD and LOQ)
Imp.2 (1.4 g, 5.9 mmol) was dissolved in methylene chloride(30 mL) inside a 250 mL RBF, and the solution was supplemented with sodium cyanate(1.42 g,21.8 mmol)upon stirring,cooled to 0°C,and dropwise treated with methanesulfonic acid (2.36 g, 3.5 mL,24.6 mmol).After the addition was completed,the reaction mixture was warmed to room temperature and stirred overnight at this temperature.The reaction was monitored using HPLC.After the reaction was complete, the solvent was removed under reduced pressure,the residue was dissolved in H2O(50 mL),and the mixture was sonicated for 10 min to give a white solid that was filtered off,dried(1.25 g),treated with refluxing acetonitrile for 1 h,filtered,and dried (690 mg).The dried solid was treated with refluxing ethyl acetate:methanol(9:1,V/V,30 mL)for 1 h,filtered,and dried to afford imp.7(550 mg,33.3% yield,97.5% purity by HPLC)as a white solid.1H NMR(400 MHz,DMSO-d6):δ/ppm=9.89(s,1H),7.29-7.21(m,7H),5.98(s,1H),5.49(s,2H),3.99-3.87(m,3H),2.75(d,J=28.6 Hz,2H);13C NMR (101 MHz, DMSO-d6):δ/ppm = 158.1 (C=O),154.4 (C=O),153.6(C=O),138.4(C,Ar),129.2(2CH,Ar),128.3(2CH,Ar),126.2(CH,Ar), 66.2 (OCH2), 49.5 (NCH), 37.1 (PhCH2); HRMS (ESI+): calcd for C12H17N4O4+:281.12443[M+H]+;found 281.1264.
2.6.8.(R)-5-Benzylimidazolidine-2,4-dione (imp.8)
D-phenylalaninol methyl ester(1.0 g,5.6 mmol)was dissolved in water(20.0 mL)inside a 100 mL RBF,and the reaction mixture was sequentially treated with a solution of sodium carbonate (0.5 g,4.7 mmol) in water (5.0 mL) and potassium cyanate (1.0 g,12.3 mmol),stirred at 80°C for 1.5 h, and extracted with ethyl acetate.The combined organic extracts were dried over anhydrous sodium sulfate and concentrated under reduced pressure to give imp.8(0.44 g, 35% yield, 99.7% purity by HPLC) as a white solid.1H NMR(400 MHz,DMSO-d6):δ/ppm=10.42(s,NH),7.91(s,NH),7.29-7.17(m, 5H), 4.32 (t,1H, J = 4.8 Hz), 3.14-3.00 (m, 2H) ppm;13C NMR(101 MHz,DMSO-d6):δ/ppm= 175.3(CH2C=O),157.3(NHC=ONH),135.8 (C, Ar),129.9 (2CH, Ar),128.2 (2CH, Ar),126.8 (CH, Ar), 58.55(CHNH), 36.59 (PhCH2); HRMS (ESI+): calcd for C10H11N2O2+:191.08150[M+H]+;found 191.0823.
3.Results and discussion
3.1.Impurity detection and identification
Fig.4 presents a representative analytical HPLC chromatogram of solriamfetol spiked with eight impurities that had been detected in crude solriamfetol during process development studies and identified by LC-MS.Consequently, these impurities were herein synthesized in quantities sufficient for full characterization and analytical (HPLC) method validation.All synthesized impurities were co-injected with solriamfetol to confirm their identity based on retention time matching (Fig.4).The high-performance liquid chromatography with ultraviolet detection (HPLC-UV) chromatograms of each impurity separately are given in Figs.S2-S9.Figs.S10-S17, S18-S33, and S34-S41 present the Fourier transform infrared spectra, original1H and13C NMR spectra, and highresolution mass spectra of imps.1-8, respectively.The assignments corresponding to1H and13C NMR spectra are presented in Section 2.All spectral data confirmed the structures of the synthesized impurities.

Fig.4 .Results of high-performance liquid chromatography with ultraviolet(HPLC-UV)analysis.Chromatograms of a 1 mg/mL solution of solriamfetol spiked with all impurities (1 μg/mL each) (a) and a blank solution (b).API: active pharmaceutical ingredient.
3.2.Elucidation of impurity structure and formation mechanism
The positive ionization mode mass spectrum of the impurity at a relative retention time(RRT)(relative to API peak)of 1.6 showed a molecular ion ([M+H]+, m/z = 195.1123) corresponding to a molecular weight of 194 amu,which agreed with the structure of imp.1.This impurity was prepared by heating a basic aqueous solution of I to 80-90°C in the presence of potassium cyanate and was fully characterized and standardized for advanced analytical studies.For all prepared impurities, the used precursors are enantiomerically pure isomers,and given that the reactions do not involve the chiral centers,the products(i.e.,impurities)are pure isomers,as indicated by their names in Table 1.
The impurity at RRT 2.1 was observed in crude solriamfetol during process development studies.The corresponding positive ionization mode mass spectrum revealed a molecular ion([M+H]+,m/z = 191.0823) corresponding to a molecular weight of 190 amu,which agreed with the structure of imp.8.To validate this assignment, we synthesized imp.8 by reacting D-phenylalaninol methyl ester (XIII) with potassium cyanate in the presence of sodium carbonate and the result showed that its RRT matched that of the corresponding impurity found in the API.Imp.8 was purified,characterized, and scaled up for analytical studies.
10、产品静放72小时:静置过程中定期打开套管升高座、联管等上部的放气塞进行放气,待油溢出时关闭塞子。
The positive ionization mode mass spectrum of the impurity with RRT 2.7 revealed a molecular ion ([M+H]+, m/z = 238.1189)corresponding to a molecular weight of 238 amu, which was slightly higher than that of solriamfetol and agreed with the structure of imp.2.Substantial amounts of imp.2 were obtained by reacting I with sodium cyanate in the presence of methanesulfonic acid.This transformation was assumed to proceed via the formation of solriamfetol as an intermediate.
The positive ionization mode mass spectrum of the impurity with RRT 4.1 showed a molecular ion ([M+H]+, m/z = 238.1185)corresponding to a molecular weight of 237 amu, which agreed with the structure of imp.6.This impurity was synthesized by reacting XII with I.
1.1 研究对象 收集2013年1月-2017年1月上海市儿童医院诊治或远程会诊的藏族(44例)和汉族(39例)DDH患儿病例资料作为研究对象。所有病例均经X片与CT诊断符合DDH诊断标准, 均为全脱位或半脱位伴髋臼发育不良者,双膝踝关节活动良好,无内外翻畸形、双足无内外翻及马蹄畸形。排除病理性髋脱位,神经肌肉疾病所致髋脱位。
The positive ionization mode ESI mass spectrum of the impurity with RRT 4.5 showed a molecular ion ([M+H]+, m/z = 281.1264)corresponding to a molecular weight of 280 amu, which agreed with the structure of imp.7.This impurity was synthesized by reacting imp.2 with excess cyanate and methanesulfonic acid.
The positive ionization mode mass spectrum of the impurity with RRT 5.5 showed a molecular ion ([M+H]+, m/z = 178.0857)corresponding to a molecular weight of 177 amu, which was 17 amu lower than that of solriamfetol and complied with the structure of imp.3.This impurity was synthesized in five steps starting from carbamate II.In the first step, the carboxylic acid group was converted to the corresponding acid chloride, which was then reacted with methanol to form the corresponding ester III.The subsequent reduction of III with sodium borohydride yielded the primary alcohol IV, which was then reacted with methyl chloroformate in the presence of pyridine to give V.After that, V was hydrolyzed to afford IV, which was catalytically hydrogenated to give the desired 5-membered ring product, i.e., imp.3.
The positive ionization mode mass spectrum of the impurity with RRT 8.7 showed a molecular ion ([M+H]+, m/z = 299.1730)corresponding to a molecular weight of 298 amu, which agreed with the structure of imp.4.This impurity was synthesized by reacting carbamate II with VII in the presence of methyl chloroformate and N-methylmorpholine to give VIII, which was then catalytically hydrogenated to produce imp.4 in quantitative yield.
603 术前糖类抗原 19-9 水平评估不同甲胎蛋白水平肝细胞癌肝切除术后患者的预后 李风伟,邹奇飞,薛 辉,项红军,夏 勇,李 俊,阎振林,沈 锋,王 葵
The positive ionization mode mass spectrum of the impurity with RRT 9.0 showed a molecular ion ([M+H]+, m/z = 342.1808)corresponding to a molecular weight of 298 amu, which agreed with the structure of imp.5.This impurity was synthesized in three steps.VII was carbamoylated to produce IX, which was subsequently reacted with II and methyl chloroformate in the presence of N-methylmorpholine to give X.Finally, dehydrogenation followed by salt formation afforded imp.5.
3.3.Proposed formation mechanisms of the identified impurities
The N-carbamoylation-derived imp.1 is a process-related impurity that can be formed during API manufacturing.In the API production process disclosed in patent US5955499A [6], a carbamate moiety is introduced to protect the amino group of D-phenylalaninol.Traces of unprotected D-phenylalaninol may react with phosgene and ammonia at the amino group (and not at the hydroxyl group) to produce imp.1.In patent WO202150067A2, this impurity was denoted as a urea impurity and was suggested to formvia the migration of the carbamoyl group from the oxygen to the nitrogen of the API under basic conditions [18].
从1958到2018,北京表用一甲子的时间磨砺高复杂制表技艺,揉和古老东方美学艺术,为狗年特别款腕表赋予更多纪念意义。
The bicarbamate imp.2 was presumably formed through the reaction of N-unprotected API traces with phosgene and ammonia according to the process disclosed in patent US5955499A [6].
Patent WO202150067A2 denoted the cyclic imp.3 as a cycle impurity and suggested that it was formed by the loss of ammonia from the API at low pH followed by the intermolecular formation of a five-membered cycle [18].
According to the API production process disclosed in patent US5955499A[6],phosgene can catalyze the conversion of traces of D-phenylalanine in D-phenylalaninol to the corresponding acid chloride, which directly reacts with the amino functional group of phenylalaninol to form imp.4.
If imp.4 is present in the API, it can react with phosgene and ammonia used for carbamoylation to give imp.5.Fig.S42 summarizes the proposed pathways for the formation of imps.1-5 in solriamfetol.
The N-dicarbonimidic diamide imp.6 can be formed under conditions similar to those affording imp.1,that is,the amino group of N-unprotected D-phenylalaninol traces can repeatedly react with phosgene and ammonia to produce imp.6.
The O-dicarbonimidic diamide imp.7 can be formed under conditions similar to those affording imp.2 via the further carbamoylation of the carbamate imp.7 at its NH2group.
监理单位需要加强施工人员内的施工监督力度,在开展防水工程关键部位施工阶段,需要进行旁站、监督,对施工过程、成品保护的逐项检查,并对隐蔽工程桩做好验收记录、检查记录、分项检查记录,要求每道工序必须经过隐蔽检查,签署认可之后,才能开展后续施工[2]。
The D-phenylalanine-cyclized imp.8 can be formed in the presence of phosgene and ammonia via the intramolecular carbamoylation of the acid chloride traces of D-phenylalanine found in Dphenylalaninol.Fig.S43 summarizes the proposed pathways for the formation of imps.6-8 in solriamfetol.
3.4.Validation of the HPLC method
The chromatographic method was developed by testing different stationary phases and mobile phase compositions and validated according to the ICH Q2 (R1) guidelines [22] for system suitability, selectivity, linearity, recovery, precision, limit of quantitation (LOD), and limit of detection (LOQ).The concentration of the solriamfetol test solution equaled 1 mg/mL, and the impurity concentration was set to 0.1% of this value, i.e.,1 μg/mL.
3.4.1.Selectivity
样品破碎和锆石挑选由廊坊峰之源矿物分选技术服务公司完成。样品经破碎后,人工淘洗保留重砂部分。对重砂部分进行电磁分选和重液分选,得到一定纯度的锆石样品,然后在双目镜下人工挑选出晶形完好、透明度高、无裂纹的锆石晶体。制靶时,将待测样品与标准样品TEM同时放置。磨蚀和抛光树脂靶,直至锆石核心部位暴露再进行阴极发光显微照相,阴极发光图像在中国地质科学院矿产资源研究所电子探针实验室拍摄,并结合透射光和反射光图像观察锆石内部结构。
两旁是巨大的成堆的花篮,头顶的吊灯晃得人头晕目眩。当然都是虚拟零件的成果。排着队进入会场,同事们兴高采烈,眼角眉梢抑制不住喜悦之情。她也心潮澎湃难以自已,在乱哄哄的队伍中拥挤着尖叫着。年终奖的评比即将公示,此时高兴才是应该的主情绪,热烈的反应才是好员工的证明。
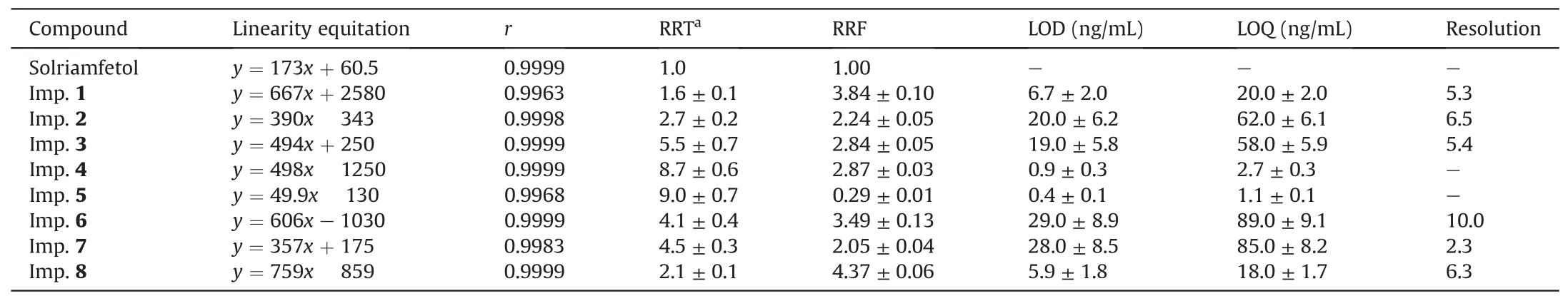
Table 2 Results of linearity,limit of quantitation(LOD),limit of detection(LOQ),relative retention time(RRT),relative response factor(RRF),and resolution evaluation obtained for the high-performance liquid chromatography (HPLC) analysis method (n = 3).
The stability-indicating power of the developed method was tested by a stress testing (also called forced degradation) study according to ICH guidelines for stability (Q1A).The results(Table S1) indicate that solriamfetol was significantly degraded under basic conditions with a total degradation degree of 23%,while the degradation in response to acidic,oxidative,and thermal stresses was moderate (4.9%, 7.3%, and 2.0%, respectively).In all stress conditions, proper mass balance, calculated by adding the values of % assay to the % degradation, was achieved (>95% in all conditions).The mass balance results prove the selectivity and the stability-indicating power of the developed method as the loss of the drug substance accompanied by almost the same extent of degradation products peaks.
3.4.2.System suitability
System suitability was evaluated by injecting five replicates of a 1 mg/mL solriamfetol solution spiked with all impurities (1 μg/mL each).The relative standard deviations (% RSDs) of peak areas and retention times ranged from 0.26% to 0.91% and were therefore less than 2.0%,the generally accepted criterion.The resolution between adjacent analyte peaks (2.3-10.0,Table 2)exceeded the minimum value (~1.5) required for baseline separation.
两组患者术后均于24 h内控制排便,术后5~7 d常规应用抗生素抗感染,观察切口变化,进行对症、支持治疗,同时保持切口干净、干燥,每天换药1次,术后7~10 d拆线。
3.4.3.Linearity, range, accuracy, precision, and robustness
合理评估国家或地区的工业化发展水平,准确判断其所处工业化发展阶段,对把握产业发展趋势和制定更好的产业发展政策具有现实意义。青岛市作为国家设立的中心城市之一,同时作为山东省经济实力最强的城市,有责任与能力带动山东半岛的经济走向更强。因此,为进一步推动青岛市经济发展,发挥其应有的作用,有必要对青岛市的工业化发展水平进行测度与分析。
Linearity was studied at 50-3000 ng/mL using nine different concentrations (each prepared in triplicate).The average peak area was plotted versus concentration, and the plot was fitted using the least squares method to obtain the corresponding linearity equation and correlation coefficient (r).The above range corresponds to 0.003%-0.3% of the nominal concentrationof solriamfetol(1mg/mL).The obtained r values(0.9963-0.9999,Table 2)indicated good linear correlation.
Accuracy and precision were studied by spiking 1 mg/mL solriamfetol with impurities at individual impurity concentrations of 500, 1000, and 1500 ng/mL and calculating % recovery using impurity standards of the same concentration.Accuracy was investigated in terms of % recovery for triplicate samples at each level.The results(86.28%-111.1%,Table 3)indicated proper analyte recovery,as the numbers were within the accepted range of 85%-115%.Precision was studied by preparing six replicates for each level.The% RSD was in the range of 0.18%-3.02% (Table 3), less than the generally accepted limit (10%).

Table 3 Result of recovery and precision evaluation obtained for the high-performance liquid chromatography (HPLC) analysis method.
2.6.7.(2 R)-2-(carbamoylamino)-3-phenylpropyl N-carbamoyl carbamate (imp.7)
LODs and LOQs were calculated from the slope (b) and the standard deviation (SD)of the y-intercept of the regression line at low analyte concentrations(50-150 ng/mL)as LOD=3.3SD/b and LOQ = 10SD/b.The obtained LODs and LOQs were in the ranges of 0.4-29.0 ng/mL and 1.1-89.0 ng/mL, respectively (Table 2).These values were less than 0.003% (LOD) and 0.009% (LOQ) of the nominal concentration of solriamfetol.The value of 0.009% obtained for LOQs was much less than the ICH guideline Q3A-stipulated threshold of 0.05% [10].
Relative response factors(RRFs)were calculated from linear fits as the ratio of the slope observed for a given impurity to the slope observed for solriamfetol and can be used to calculate impurity concentrations when no standards are available.
Thus,the developed method was concluded to be selective,precise,accurate,and suitable for the assaying of solriamfetol batches.
4.Conclusion
Eight process-related solriamfetol impurities were identified,synthesized,and characterized,and plausible mechanisms of their formation were proposed to shed light on the critical steps of API synthesis.Impurity structures were elucidated using1H and13C NMR spectroscopy, infrared spectroscopy, and LC-MS.This characterization resulted in compliance with regulatory requirements,and the prepared impurity standards were used to develop and validate a chromatographic method of impurity analysis and thus enable efficient solriamfetol quality control in industrial settings.
CRediT author statement
Nafisah Al-Rifai: Conceptualization, Methodology, Writing -Original draft preparation;Anas Alshishani:Validation,Resources,Writing - Original draft preparation;Fouad Darras: Supervision,Project administration;Ola Taha: Investigation.Shereen Abu-Jalloud: Investigation;Lena Shaghlil: Validation, Investigation;Yousef Al-Ebini: Writing - Reviewing and Editing.
市售纯牛奶中,按比例加入6%的白砂糖,95℃杀菌5 min,冷却到43℃待用。将活化后的菌株按体积分数1%的比例接种于杀菌处理的纯牛奶中,43℃发酵,跟踪酸度变化,当酸度达到70°T,终止发酵,置于4℃冰箱中冷却后熟。酸奶冷藏过夜后取出,待酸奶温度恢复到10~15℃时,选取10名食品专业人员从色泽、滋气味和组织状态等方面进行感官评价[7]。
Declaration of competing interest
The authors declare that there are no conflicts of interest.
Acknowledgments
This research was funded by the Deanship of Scientific Research at the German-Jordanian University and the Deanship of Scientific Research at Zarqa University.The graphical abstract was created with BioRender software.
Appendix A.Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jpha.2023.02.012.
猜你喜欢
军事文摘(2020年24期)2020-02-06
现代营销(创富信息版)(2018年6期)2018-09-05
现代营销(创富信息版)(2018年9期)2018-09-03
现代营销(创富信息版)(2018年10期)2018-02-21
现代营销(创富信息版)(2018年7期)2018-02-20
中国宝玉石(2016年2期)2016-10-14
新疆地质(2015年3期)2015-12-10
华北地质(2015年3期)2015-12-07
航天器工程(2014年5期)2014-03-11
中国环境科学(2014年4期)2014-02-02
 Journal of Pharmaceutical Analysis2023年4期
Journal of Pharmaceutical Analysis2023年4期
- Journal of Pharmaceutical Analysis的其它文章
- Neutrophil elastase: From mechanisms to therapeutic potential
- Development of a CLDN18.2-targeting immuno-PET probe for non-invasive imaging in gastrointestinal tumors
- High-throughput transcriptional profiling of perturbations by Panax ginseng saponins and Panax notoginseng saponins using TCM-seq
- A chiral metal-organic framework {(HQA)(ZnCl2)(2.5H2O)}n for the enantioseparation of chiral amino acids and drugs
- Recent applications and chiral separation development based on stationary phases in open tubular capillary electrochromatography(2019-2022)
- Quantitative characterization of cell physiological state based on dynamical cell mechanics for drug efficacy indication