
Obesity and cancer stem cells: Roles in cancer initiation,progression and therapy resistance
2023-05-25 09:24WenJieXieJianLi
World Journal of Stem Cells 2023年4期
Wen-Jie Xie, Jian Li
Wen-Jie Xie, Jian Li, Department of General Surgery, The Third Hospital of Mianyang, Sichuan Mental Health Center, Mianyang 621000, Sichuan Province, China
Abstract Obesity, the global pandemic since industrialization, is the number one lifestylerelated risk factor for premature death, which increases the incidence and mortality of various diseases and conditions, including cancer. In recent years, the theory of cancer stem cells (CSCs), which have the capacity for self-renewal,metastasis and treatment resistance, has been bolstered by increasing evidence.However, research on how obesity affects CSCs to facilitate cancer initiation,progression and therapy resistance is still in its infancy, although evidence has already begun to accumulate. Regarding the ever-increasing burden of obesity and obesity-related cancer, it is pertinent to summarize evidence about the effects of obesity on CSCs, as elucidating these effects will contribute to the improvement in the management of obesity-related cancers. In this review, we discuss the association between obesity and CSCs, with a particular focus on how obesity promotes cancer initiation, progression and therapy resistance through CSCs and the mechanisms underlying these effects. In addition, the prospect of preventing cancer and targeting the mechanisms linking obesity and CSCs to reduce cancer risk or to improve the survival of patients with cancer is considered.
Key Words: Obesity; High-fat diet; Cancer stem cells; Carcinogenesis; Metastasis
INTRODUCTION
For millions of years, humans and their predecessors have evolved under the pressure of undernutrition, which selects a genotype that enables overeating, low energy expenditure, a high degree of calorie absorption and efficient energy storage in adipose tissue[1]. Therefore, with the development of the social economy in the past few decades, overnutrition and an increasingly sedentary lifestyle tip the balance from a few calories consumed but more expended to more calories consumed but little expended, leading to the pandemic of excess body weight, which is mainly measured by body mass index (BMI). Over the past four decades, the prevalence of overweight and obesity has nearly tripled globally. Between 1975 and 2016, the worldwide prevalence of obesity increased from less than 1% to 6%-8% among children, from 3% to more than 11% among men and from 6% to 15% among women[2].Based on data from the Global Burden of Disease (GBD) 2015, overweight or obesity affects over 2.1 billion people, or nearly 30% of the global population[3]. Obesity was estimated to increase the economic burden by approximately 2 trillion United States dollars, or 2.8% of the global gross domestic product, and to lead to the loss of an estimated 5-20 years of life expectancy, representing one of the most serious unmet public health challenges of the 21stcentury[4-6].
Malignancy, a set of diseases caused by the interplay between genetic and environmental or behavioral factors, ranks as the third leading cause of premature death and disability attributable to excess body weight worldwide following cardiovascular disease and type 2 diabetes mellitus[7]. Recent studies have demonstrated that excess body weight is associated with higher risks of several types of cancer, including esophageal adenocarcinoma, multiple myeloma, and cancers of the gastric cardia,colon, rectum, biliary tract system, pancreas, breast, endometrium, ovary, and kidney[8]. In 2019, the estimated number of high BMI-related cancer cases accounted for 4.59% and 4.45% of all cancer-cause deaths and disability-adjusted life years, respectively[9]. Obesity can not only increase the risk of tumorigenesis but also promote the progression and metastasis of developed cancer and can affect the therapeutic efficacy and survival of patients with cancer[10].
Regarding the altered biological processes that occur in the context of obesity that contribute to cancer, the majority of studies have focused on common themes, including inflammation, hypoxia,angiogenesis and altered energy metabolism, which influence the proliferation and survival of cancer cells[10]. However, in recent years, emerging challenges in cancer management have promoted the proposal of many theories to explain the initiation and progression of cancer; one of them is the hypothesis of cancer stem cells (CSCs), which has been bolstered by an accumulating body of evidence[11]. CSCs, also referred to as treatment-refractory, tumor-initiating cells, constitute a small subpopulation of cancer cells within tumors capable of self-renewal, which can divide and differentiate into various tumor cell types (intratumoral heterogeneity). They can secrete antiapoptotic factors, undergo epithelial-to-mesenchymal transition (EMT), and display a higher performance of drug efflux pumps.Therefore, CSCs are preferentially aggressive and pose a high risk of therapy resistance and disease relapse[11]. With the rapidly increasing incidence of cancer attributable to obesity, a better understanding of the roles of obesity in CSC biology is of paramount significance. However, research in this area is in its infancy. In this review, we discuss the association between obesity and CSCs, with a particular focus on how obesity promotes cancer initiation, progression and therapy resistance through CSCs and the mechanisms underlying these effects. In addition, the prospect of prevention and targeting mechanisms linking obesity and CSCs to reduce cancer risk or to improve the survival of patients with cancer is considered.
OBESITY AND CSCS IN CANCER INITIATION
In the context of an increase in the global prevalence of obesity, large-scale epidemiological studies have demonstrated a compelling increased risk of tumorigenesis in individuals with obesity, and several landmark studies have summarized this evidence. To evaluate the strength and validity of the evidence for the association between adiposity and the risk of developing or dying from cancer, an umbrella review of the literature comprising 204 meta-analyses of large studies with limited heterogeneity or evidence of bias was published in 2017, which concluded that the associations for 11 cancers were supported by strong evidence, while others could be genuine, but substantial uncertainty remains[8]. In 2016, data from a meta-analysis reported by the International Agency for Research on Cancer supported relative risks of 1.5 to 1.8 in obesity for these tumor sites[12]. In GBD 2019, 13 cancer types were also found to be affected by a high BMI[9]. Although some inconsistencies in cancer types contributing to obesity were reported across these studies, a consistent and compelling association has been demonstrated in many cancer types, including esophageal adenocarcinoma, multiple myeloma, and cancers of the gastric cardia, colon, rectum, biliary tract system, pancreas, breast, endometrium, ovary,and kidney (Figure 1). In the majority of these cancers, the CSC theory has been established in tumorigenesis. For example, in the intestine, inactivation of the adenomatous polyposis coli (APC) gene can lead to the rapid and lethal generation of adenomas in intestinal stem cells (ISCs) but not in non-stem cells[13]. Breast cancer was found to originate from a rare population of mammary gland progenitor cells, the depletion of which significantly impaired tumor growth[14]. Therefore, to increase the risk of these cancers, obesity may disturb the normal biology of stem/progenitor cells residing in these tissues,which is conducive to their transformation.
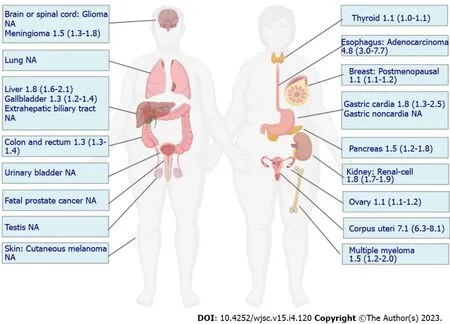
Figure 1 Relative risk of individual cancers at high body mass index.
Except for compelling epidemiological evidence, no attempts have been made to investigate the biology of cancer-related adult stem cells in populations with obesity. Multiple animal models have been developed to recapitulate the effects of obesity or a pro-obesity diet on the initiation of cancer and have suggested that a high-fat diet (HFD) can promote tumorigenesis in the colorectum, prostate and liver[15-17]. However, the cellular origin of cancer was not defined in these studies. In recent years,research revealing the links between obesity or HFD and adult stem cells has increased (Table 1)[18-28].Although discrepancies exist, the majority of these studies reported one or more of the following findings: Obesity or a HFD increases the depth or number of crypts in the intestine; non-stem cell progenitors in a HFD setting acquire stem cell attributes; the number and the capacity to form organoids of stem cells or progenitor cells are increased by a HFD; stem cells undergo autonomous changes in response to a HFD that poise them for niche-independent growth. Although studies exploring the initiation of carcinogenesis from these stem cells are very limited, the alterations in stem cells reported in these studies can predispose them to transformation. First, obesity or a HFD expands the pool of cells- both stem cells and progenitor cells - that can serve as the cellular origin of nascent cancers. Second,stem cells from mice on a HFD become functionally uncoupled from their niche in the organoid assay andin vivo, consistent with the hallmarks of cancer cells. Third, several studies have shown the possible links between these perturbations and tumorigenesis. For instance, when injected with azoxymethane,aberrant crypt foci (ACF), an early-appearing lesion of colon carcinogenesis, were increased in male mice fed a HFD[21]. In another study, more spontaneous intestinal low-grade adenomas and carcinomas were observed in HFD-fed mice than in standard diet-fed mice[19].

Table 1 Murine models of high-fat diet-induced obesity and cancer stem cells
At present, elucidating how obesity and a pro-obesity diet contribute to the cellular origin of cancer in the intestine is the central focus of research, and data on stem cells in other tissues are very limited.Several reasons can explain such a tissue preference for studying the impact of obesity and HFD physiology on the initiation of cancer. First, robust epidemiological evidence has been accumulated for the increased risk of colon cancer in obese populations, and a better understanding of the altered biology of ISCs that occurs in the context of obesity will provide immeasurable public health benefits[10]. Second, the stem cell theory advanced most rapidly in ISCs, from which the histological architecture of the intestine has been well established[29]. Third, ISCs reside in the base of the intestinal crypt and directly interact with luminal nutrients, bacteria, and other intraepithelial and subepithelial cells, making the intestine an ideal system for studying the pathophysiological changes on a HFD[29].Finally, the natural orifice of the intestine makes in situ manipulations for tumor induction or diagnostic tests easier than otherin vivocancer models[30]. Despite all of this, as consistent and compelling associations have been demonstrated between obesity and more than 10 cancer types, elucidating how stem cells in tissues other than the intestine are perturbed by obesity or HFD holds the same importance.Currently, the majority of obesity models are induced by HFD; however, other dietary patterns, such as a high-sugar diet and Western-styled diet, have also been shown to be obesogenic, and the effects of these dietary patterns on the initiation of cancer warrant further studies. Furthermore, the alterations in stem cell biology in tissues with increased cancer incidence warrant further investigation in obese human beings, not just in animals.
OBESITY AND CSCS IN CANCER PROGRESSION AND THERAPY RESISTANCE
In addition to promoting tumorigenesis, obesity might also promote the progression of established cancers, affect the efficacy of present forefront antitumor therapies and shorten the survival of patients with cancer. For instance, a meta-analysis including 86490 patients treated for clinically localized prostate cancer showed a moderate and consistent relationship between obesity and biochemical recurrence, and there was a 10% increase in biochemical recurrence per 5 kg/m2increase in BMI[31]. In the Carolina Breast Cancer Study phase 3, a high waist-to-hip ratio was found to be associated with a high risk of metastasis[32]. Poor survival was also reported in overweight or obese patients with colorectal, endometrial and breast cancer[33-35]. In addition to the increased likelihood of recurrence,the poor prognosis of obese patients with cancer also results from the reduction in the efficacy of antitumor therapies[36]. Mechanistically, the link between obesity and increased recurrence, therapy resistance and poor survival is likely multifactorial, with some differences related to more advanced stages being attributed to reduced participation in routine screening or the systemic effects of obesity on drug pharmacokinetics and metabolism[37,38]. In addition to these explanations, emerging evidence has shown that the activation of stem cell programs in cancers can lead to progression, metastatic growth and therapy resistance[11].
The key roles of obesity in the activation of stem cell programs have attracted much attention in recent years; however, as in studies on the effects of obesity on cancer-initiating cells, the promotion effects of obesity on cancer through CSCs are also mainly limited to animal models, which are utilized to investigate how specific obesity-related factors induce the stemness of cancer cells. Knowledge about CSCs in obese patients with cancer is still not clear. For example, obesity increases inflammation in the tumor microenvironment (TME) through local and systemic adipokines, proinflammatory cytokines or hormones, which modulate the stemness of cancer cells[39]. In a mouse model of hepatocellular carcinoma, diet-induced obesity increased inflammatory signalingviaSTAT3, and this finding was associated with larger tumors with a cancer-stem-cell-like phenotype[40]. Prolonged culture of breast cancer cells, which developed from a fat-rich environment, with adipocytes increased the proportion of cells expressing stem-like markersin vitroand the abundance of cancer cells with metastatic potentialin vivo[41]. Regarding the involvement of CSCs in therapy resistance, leptin was found to interfere with the efficacy of 5-fluorouracil (5-FU) in colon tumor stem cells by increasing cell viability and reducing 5-FU-induced DNA damage[42].
EMT is a reversible cellular process during which epithelial cells transiently acquire mesenchymal phenotypes, such as an elongated, fibroblast-like morphology as well as an increased capacity for migration and invasion[43]. It is now widely accepted that EMT has well-established roles in cancer metastasis[43]. In the majority of carcinomas, only CSCs exhibit aspects of EMT-program activation[44].Various extracellular stimuli, including obesity-related factors, have been implicated in the induction of EMT programs. For instance, esophageal cancer cells cocultured with visceral adipose tissue taken from obese patients resulted in the induced expression of genes involved in EMT, which was also noted in tumor biopsies from obese patients[45]. Cytokines and growth factors released by adipose stem cells(ASCs) can induce EMT-like changes in various cancer cells[10]. The adipokine leptin has also been found to activate EMT programs to enhance the proliferation and metastasis of breast cancer cells[46].Therefore, obesity can propel primary tumor cells toward EMT events, leading to malignant progression.
THE LINKS BETWEEN OBESITY AND CSCS
As discussed above, CSCs participate in every step of tumorigenesis promoted by obesity.Understanding the key links between obesity and CSCs, therefore, offers important potential to decrease the incidence and improve the outcomes of obese patients with cancer. Several main factors are considered to connect obesity and cancer: Components of pro-obesity diets, metabolic and hormonal alterations associated with obesity, dysfunctional adipose tissue in the TME, low-grade obesity-related inflammation, self-renewal and stemness pathways, and microbiome dysbiosis. Each of these factors is intimately linked to and cross-talks with each other. For example, fatty acids in pro-obesity diets accumulate in adipocytes, leading to expansion and dysfunction of adipose tissue, which is intimately linked to endocrine and paracrine dysregulation, such as increased circulating insulin, insulin-like growth factor-1 (IGF-1) and leptin. All of these alterations activate and maintain a prolonged low-grade inflammatory state, predisposing individuals with obesity to an increased cancer risk and poor outcomes[47-49]. Although these factors were mainly investigated in nonspecific conditions, their involvement in CSC biology is also beginning to accumulate evidence (Figure 2).
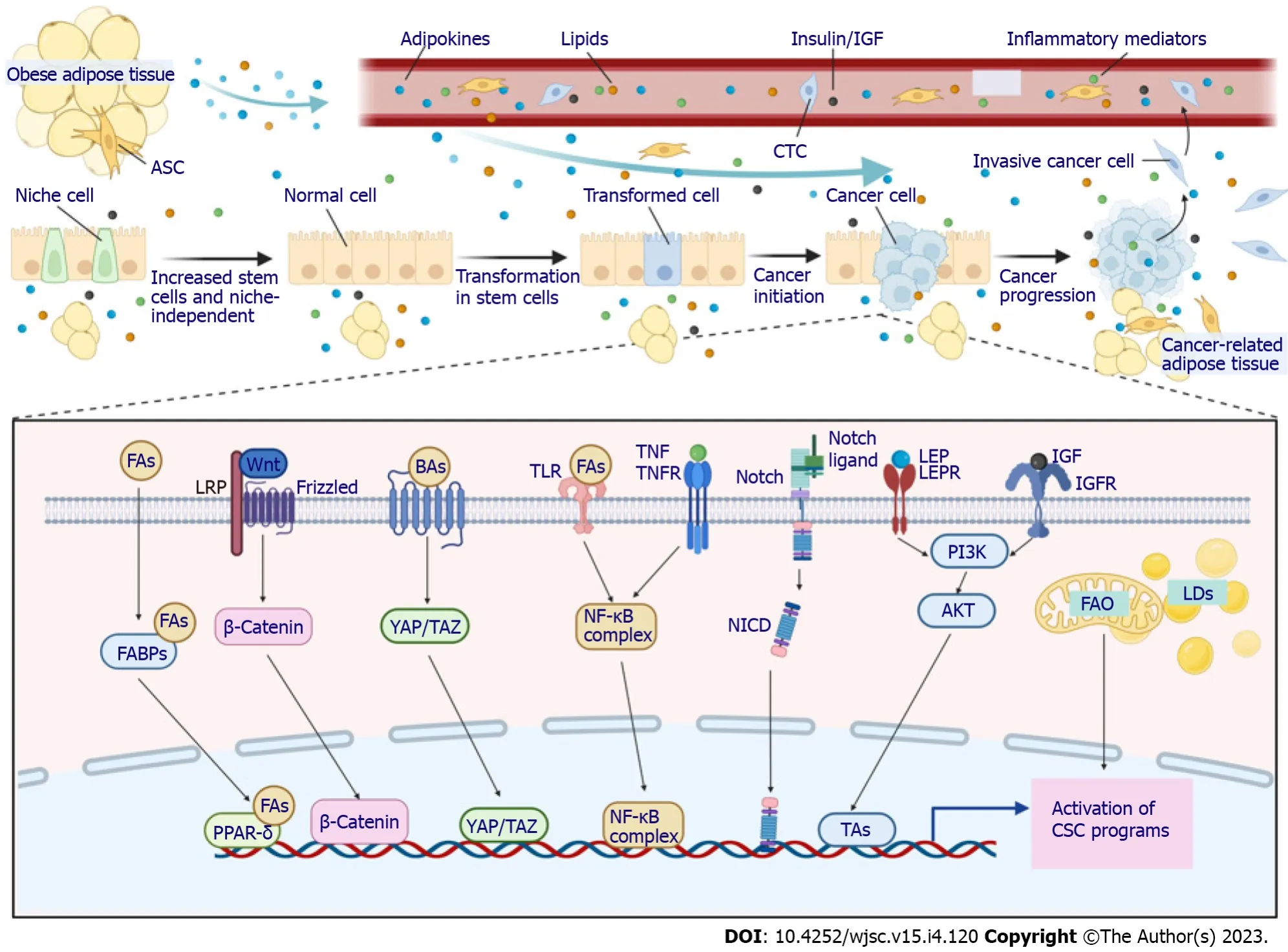
Figure 2 Mechanisms linking obesity and cancer stem cells.
Components of pro-obesity diets
Although the mechanistic links are not completely understood, nutrient-sensing signaling activated by components of a pro-obesity diet has been shown to influence stem cell behavior and tumorigenesis.This was also indirectly suggested in a leptin-receptor-deficient (db/db) mouse model, which becomes obese on control diets and does not rely on a HFD. Indb/dbmice, the number of ISCs was reduced, while ISC function was not affected, highlighting that components of a pro-obesity diet can regulate stem cells independently of obesity[19]. As early as 1989, Blakeboroughet al[50] reported that diets high in fat support aBacteroides-dominated colonic microflora and increase the excretion of secondary bile acids to augment free radical production, which may overcome the antioxidant defense mechanisms of stem cells, causing DNA damage, tumorigenesis and proliferation of transformed stem cells. Since then,numerous studies have suggested for decades that a pro-obesity diet engages many diverse pathways in stem cells in various tissues that collectively contribute to tumorigenesis. For instance, a HFD increases leucine-rich repeat-containing G protein-coupled receptor 5 (Lgr5) expression and promotes tumor growth in a xenograft model independent of obesity. Mechanistically, dietary fats stimulate vitamin Abound serum retinol-binding protein 4 and retinoic acid 6, which are implicated in colon stem cell selfrenewal, to activate the JAK-STAT3 pathway and boost Lgr5 expression and the tumorigenicity of ISCs[51].
The representative components of a pro-obesity diet, that is, fatty acids such as palmitic acid or oleic acid, were found to enhance the number and self-renewal potential of ISCs and to permit organoid body formation without supporting signaling from their niche cells in a well-designed study[19]. The molecular mechanism by which fatty acids deregulate ISCs to promote tumorigenesis was also delineated in this study. It was proposed that fatty acids can be transported to the nucleus by fatty acidbinding proteins or can be produced directly by lipid metabolism in the nucleus, where they increase the number and self-renewal of ISCsviaperoxisome proliferator-activated receptor δ (PPAR-δ), a nuclear receptor that senses fatty acid derivatives, the synthetic activation of which mimics both thein vivoandin vitroimpact of a HFD and fatty acid treatment[19]. In contrast, loss of PPAR-δ completely abrogated the effects of fatty acids on Lgr5+ ISC function with respect to organoid-initiating capacity[19]. The downstream signaling mediating the effects of PPAR-δ activation was attributed to WNT/Bcatenin, as demonstrated by increased B-catenin staining and upregulation of its target genes (Jag1, Jag 2 and Bmp4) in ISCs and progenitors from HFD- and PPAR-δ agonist-treated mice[19]. Free fatty acids produced by obese fat lipolysis also serve as ligands for Toll-like receptor 4 (TLR4) on cancer cells to activate nuclear factor-kappaB (NF-κB), leading to an increase in CSCs[52]. Another critical element in a pro-obesity diet that is significantly elevated in some obese individuals, cholesterol, was also demonstrated to affect stem cell function, thus promoting tumorigenesis. InDrosophila, dietary cholesterol modulated the differentiation of ISCs by stabilizing the Delta ligand and Notch extracellular domain and altering their trafficking in endosomal vesicles, while a low-sterol diet slowed the proliferation of enteroendocrine tumors initiated by Notch pathway disruption[53]. In a rodent animal model,evidence also showed that dietary cholesterol acts as a mitogen for ISCs, while disruption of cholesterol homeostasis dramatically enhances tumor formation in APCminmice[54].
Although bile acids are not elements in a pro-obesity diet, their excretion is essential for the digestion and absorption of dietary fat and is increased when a HFD is consumed. Bile acids are endogenous agonists of the G protein-coupled bile acid receptor, the activation of which augments Yes-associated protein 1 (YAP1) signaling, leading to increased stem cell number and proliferation and enhanced organoid-forming capability of ISCs[55]. In addition, bile acids, such as tauro-B-muricholic acid and deoxycholic acid, can antagonize intestinal farnesoid X receptor (FXR), a master regulator of bile acid homeostasis. Antagonizing FXR in the intestinal lumen enhances the proliferation and DNA damage of stem cells, initiating the transformation of ISCs to a malignant phenotype and promoting adenoma-toadenocarcinoma progression[56]. Conversely, selective activation of intestinal FXR by its agonist can restrict abnormal ISC growth and skew differentiation toward goblet cells, thus curtailing HFD-induced intestinal cancer progression[56].
Metabolic alterations
Aerobic glycolysis has long been viewed as the main metabolic characteristic of cancer cells. However,in recent years, CSCs have been found to be intimately dependent on lipid metabolism to maintain their self-renewal capability. Therefore, in addition to the abovementioned studies that investigated the regulation of CSC function by lipids as elements of a pro-obesity diet, numerous studies have explored the effects of lipids on CSCs at the cellular metabolism level. Metabolism of fatty acids and cholesterol,includingde novobiosynthesis, storage and fatty acid oxidation (FAO), supports the stemness, proliferation and chemotherapy resistance of CSCs[57]. Metabolic analysis demonstrated that lipid synthesis,includingde novolipid biosynthesis, lipid desaturation, and cholesterol synthesis, displays high activity in CSCs, indicating that lipid synthesis plays critical roles in stemness maintenance[58]. Human breast cancer-derived data suggest that FAO promotes cancer cell stemness and chemoresistance. Blocking FAO resensitizes them to chemotherapy and inhibits CSCs in mouse breast tumorsin vivo[59].Furthermore, cytarabine-resistant acute myeloid leukemia cells, which are enriched in leukemic stem cells, exhibited increased FAO[60]. FAO is also responsible for the stemness and chemotherapy resistance in gastric cancer induced by mesenchymal stem cells (MSCs)[61]. To meet the critical functions of lipids in CSCs, lipid droplets, organelles that store neutral lipids, accumulate and are more abundant in CSCs in numerous types of cancer[57]. Although evidence connecting lipid metabolism and CSCs is increasingly accumulating, whether obesity can augment the lipid metabolic alteration in CSCs is not clear because studies on the metabolic adaptations of CSCs in obese environments are limited.However, the incidence of hyperlipidemia is higher in obese populations, and in obese individuals,CSCs more readily reside in a fat-rich TME, which may provide more lipids to CSCs. Therefore, theoretically, lipid metabolic alterations in obesity support the stemness of cancer cells, although further studies are warranted to validate such effects.
Another common metabolic alteration of obese patients is insulin resistance, leading to increased levels of circulating insulin and IGF-1, which contribute to the increased risk and mortality of several cancers in obese individuals. Mice with diet-induced obesity exhibited increased concentrations of plasma glucose, insulin, and IGF-1, which were significantly correlated with increased proliferation and self-renewal of ISCs, as well as decreased Paneth cell numbers[26]. In addition, insulin significantly increased the capacity of organoid formationin vitro[26]. Reports have suggested that the PI3K/AKT pathway is the major contributor to the abnormal renewal of ISCs endowed by insulin/IGF-1[62].Therefore, insulin/IGF-1 signaling was suggested to mediate the effects of obesity on the function of stem cells, which is conducive to their transformation. Even insulin/IGF-1 levels in newborns are associated with the risk of future breast cancer, possibly resulting from an increased total number of stem cells[63]. Parallel to their function in normal stem cells, evidence suggests the roles of insulin/IGF-1 in cancer progenitor/stem cells from solid and hematopoietic malignancies. Insulin/IGF-1 and their receptors are overexpressed or overactivated in human thyroid, hepatic and breast CSCs and participate in the self-renewal, EMT and chemoresistance of cancer cells[64]. These emerging discoveries will undoubtedly promote renewed efforts aimed at targeting the insulin/IGF-1 system that contribute to CSC biology.
Hormonal alterations
Adipose tissue has long been viewed as an energy reservoir; however, this perspective has changed in recent years, as numerous bioactive adipokines, including more than 50 different metabolic and hormonal factors, cytokines and chemokines, were reported to be released by adipose tissue[65]. Two of the major adipose tissue-derived hormones are leptin and adiponectin, which have opposite effects. In contrast to lean adipose tissue, which mainly secretes the antimitogenic adipokine adiponectin in obesity, increased preadipocytes yield high levels of leptin, which has proangiogenic and promitogenic effects[66,67].
Leptin acts as a growth factor for many tissues, such as the mammary gland, lung, liver, and colonic epithelium[68]. The links between leptin and CSCs have been comprehensively studied in breast cancer[69]. In a diet-induced obese mouse model, mammary epithelial polarity was disrupted, which can contribute to overactivation of the PI3K/AKT pathway downstream of the paracrine effect of leptin expressed by neighboring adipocytes. Leptin expands the pool of stem/progenitor cells in the breast epithelium and causes mitotic spindle misalignment, which is an early step in tumor initiation[70]. The leptin receptor was found to be expressed on breast CSCs, and in orthotopically transplanted breast cancer, leptin can promote CSC enrichment[71,72]. Inactivation of the leptin receptor attenuated the expression of CSC transcription factors and reduced the self-renewal of cancer cells in tumor sphere assays[71]. Leptin-mediated cancer initiation, progression and therapy resistance through CSCs in other cancers have also been extensively investigated[69]. For instance, leptin was found to initiate the early transformation of colon cancer. ACF multiplicity, as early-appearing lesions of tumorigenesis, was increased by a HFD inob/obmice or in a genetic mouse (db/db) model with leptin receptor deletion[73,74]. However, indb/dbmice fed a control diet, the function of ISCs and the activity of PPAR-δ and Wnt/B-catenin signaling were not changed, indicating that obesity and elements in a pro-obesity diet may cause different alterations in the function of stem cells[19]. In addition, no leptin receptor was found on colonic stem cells, and leptin did not increase the pool of Lgr5+ stem cells, suggesting that leptin may be dispensable in the early stages of colon carcinogenesis[22]. Collectively, these findings indicate that although the crucial role of leptin in CSCs may be affected by the cellular origin of cancer, the potential of leptin pathways in cancer initiation and progression will lead to future areas of therapeutic management.
Although data are scarce, other adipokines with altered secretion in obese adipose tissue were also shown to affect the function of CSCs. For example, DeClercqet al[22] specifically investigated the effect of a HFD on colonic stem cell maintenance during cancer initiation and found that the number of stem cells and their proliferation capacity were significantly increased, while the incidence of apoptosis was decreased. The authors proposed that these effects are the result of decreased adiponectin signaling based on the findings that the reduction in stem cell number and increase in apoptosis were diminished in organoid cultures from obese mice treated with an adiponectin receptor agonist[22]. In addition,following a decrease in adiponectin signaling, obesity can increase tumorigenesis in the intestine[22].Resistin, another adipokine, was highly associated with the transcription of genes related to CSCs in low malignant breast cancer cells and noncarcinogenic breast epithelial cells[75]. These adipokines with different effects on CSCs and their therapeutic translational potential need further research.
Dysfunctional adipose tissue in the TME
Despite the systemic effects of adipose tissue on CSCs through the secretion of circulating metabolic and hormonal factors, adipose tissue also constitutes an important part of the microenvironment of several cancers, and its dysfunction resulting from obesity is considered a critical determinant of cancer progression[76]. For example, cancer-associated adipose tissue obtained from obese patients with breast cancer was found to increase inflammatory breast cancer aggressivenessviathe regulation of CSC markers[77]. Coculture of breast cancer cells with human-derived adipocytes increased the abundance of mammosphere-forming cells and stem-like cancer cellsin vitroand increased tumor-initiating cells and metastasis in mouse models[41]. Mechanistic investigations demonstrated that immature adipocyte contact activates Src, thus promoting embryonic stem cell transcription factor upregulation, including Sox2, c-Myc, and Nanog, to mediate CSC expansion[41]. Moreover, Sox2-dependent induction of miR-302b further stimulated c-Myc and Sox2 expression and potentiated cytokine-induced CSC-like properties[41]. However, adipose tissue from different anatomical sites may have different effects on CSCs. For example, serial transplantation of pluripotent stem cells cultured in conditioned medium of breast cancer cells into mammary fat pads evoked the same features of breast cancer, while this result was perturbed following subcutaneous transplantation, indicating that mammary adipose tissue can synergize with secretory factors produced by cancer cells to transform normal cells into CSCs, while subcutaneous adipose tissue cannot[78]. Such performance differences may be caused by the various metabolic characteristics of adipose tissue from different anatomical sites determined by sex steroid hormones[79].
Among various adipose tissue cell types, ASCs are key players in adipose tissue. Under obese conditions, the biology of ASCs is dramatically altered, and ASCs can be recruited to sites of inflammation, including tumors[80]. ASCs are able to produce a large variety of circulating growth factors,cytokines and adipokines, which play important roles in CSC function. In addition to their systemic effects, ASCs represent an important cellular component in the TME. Therefore, in a breast cancer patient-derived xenograft model, cancers grown in the presence of ASCs had increased numbers of CD44+CD24- CSCs in the peripheral blood and had a higher tendency to form metastases[81]. This effect may be mediated by leptin, as the stable knockdown of leptin in obese ASCs led to a significant reduction in circulating CSCs[81]. In addition, ASCs reshape the TME and support the generation of CSCs, which are associated with radioresistance and chemotherapy resistance[82].
Low-grade obesity-related inflammation
In recent decades, the contribution of inflammation to cancer initiation, progression and therapy resistance has regained enormous interest, and the association between inflammation and CSCs has also been explored extensively[83]. Obesity has long been considered a facilitator of mild, chronic, systemic inflammation. Along with the expansion of adipose tissue in obesity, hypoxia causes adipocyte stress and malfunction, recruiting different types of immune cells[84,85]. Both adipocytes and immune cells release numerous adipokines, cytokines, chemokines and hormones, which perpetuate the inflammatory state[84,85]. Therefore, it is reasonable to speculate that obesity can affect CSCs through lowgrade inflammation. Although chronic inflammation is not induced consistently in obese mouse models,which may be related to the differences in feeding patterns and other combined interventions, current evidence indicates that inflammation might have a role in alterations in stem cells leading to tumorigenesis. Inflammatory mediators were found to be increased in the intestinal mucosa in mice with obesity or those fed a HFD, resulting from cytokine release by myofibroblasts and immune cells[86].Local inflammation has been demonstrated to expand colon cell progenitors or stem cells and to induce their proliferation in the intestine. For example, HFD-induced obesity elevated both the colonic proliferative zone and stem cell zone in a pig model, and the proliferative zone was associated with an increase in the innate inflammatory markers TLR4, NF-κB, IL-6, and lipocalin-2[87]. In addition,activation of NF-κB, the central pathway downstream of the majority of proinflammatory cytokines,following local inflammation enhanced the reprogramming of non-stem enterocytes to acquire stemcell-like properties, which expanded the pool of stem cells and generated tumor-initiating cells[88].HFD-induced obesity promoted the phosphorylation of GSK3B and then increased the nuclear translocation of B-catenin, thereby activating the expression of WNT signaling target genes[89]. These effects were diminished by the deletion of tumor necrosis factor-alpha (TNF-α), indicating the role of inflammation induced by TNF-α in colon tumorigenesis associated with obesity[89]. Another inflammatory mediator, prostaglandin E2, was also found to be elevated by HFD in the circulation or in local tissues,leading to an increased number and division rate of stem cells[22].
Other proinflammatory mediators have also been demonstrated to facilitate CSC expansion. For example, IL-6 can induce malignant features in human ductal breast carcinoma stem/progenitor cells[90]. IL-8 treatment leads to breast cancer cells partially acquiring some stem-like cell attributes, thereby increasing their aggressiveness[91]. Chemokine (C-C motif) ligand 2, derived from cancer-associated fibroblasts, stimulates the stem cell-specific, sphere-forming phenotype in breast cancer cells and CSC self-renewal[92]. Although these studies were not carried out under obese conditions, these proinflammatory mediators are consistently elevated in obese individuals and are upregulated upon contact with cancer cells. The enrichment of CSCs may be partially attributed to these cytokines, as a few studies indeed found that proinflammatory cytokines, including IL-6, IL-8 and monocyte chemoattractant protein 1, are overexpressed in cancer-associated adipose tissue from obese patients and induce the stemness of cancer cells, while such effects were not found in nonobese patients[77]. Considering the importance of localized and systemic inflammation in the induction and maintenance of stemness in cancer cells and the definite association between inflammation and obesity, elucidating how these inflammatory pathways increase the risk of cancer incidence, progression and therapy resistanceviaCSCs holds great promise to decrease the burden of cancer in obesity.
Self-renewal and stemness pathways
Stem cells are proposed to reside in a distinctive microenvironment, that is, the stem cell niche, which induces and maintains the self-renewal and differentiation of stem cells. In the TME, niche signals also play critical roles in cancer cells acquiring more stemness. Although the pathways responsible for establishing a CSC phenotype are diverse and differ among cancer entities, developmental signaling pathways, including the Notch, WNT, Hedgehog and Hippo pathways, are commonly altered in CSCs and interact with each other and with other common oncogenic signaling pathways and have key regulatory functions that support the maintenance and survival of these cells, making them prime targets for anti-CSC therapy[82]. Obesity, HFD and abnormal adipocytes may engage in these selfrenewal and stemness pathways directly or indirectly through increased local and systemic levels of many cytokines and adipokines. For example, in ISCs and progenitors from mice fed a HFD, the expression of Jag1 and Jag2, which are ligands for the Notch pathway and are normally expressed by neighboring niche cells, was increased by the activation of WNT/B-catenin, indicating that a HFD drives ISCs to niche independence[19]. Within the ISC niche, MSCs were expanded and secreted predominant levels of Wnt2b in the colon of HFD-fed mice, which promoted the growth of tumorigenic properties and accelerated the expression of CSC-related markers in colon organoids[93]. CSCs isolated from obese mice also exhibited enhanced Notch2 expression[94]. However, such direct evidence supporting the association between obesity and alterations in stemness pathways is scarce, and more studies are needed to test this model. Nonetheless, some emerging data demonstrate alterations in these stemness pathways in obesity-induced cancers. For instance, HFD consumption could upregulate the expression of B-catenin proteins in a mouse xenograft tumor model[95]. Notch signaling activity was increased in breast cancer cells following coculture with obesity-altered ASCs[96]. In addition, YAP, the major player in the Hippo pathway, dictates mitochondrial redox homeostasis to facilitate obesity-associated breast cancer progression[97]. Although the contributions of CSCs were not examined in these studies,regarding the definitive effects of these signaling pathways on CSCs, it is reasonable to speculate that the upregulated activity of these pathways in obesity may promote the progression of cancer through CSCs and that targeting these pathways may be more promising in obesity-induced cancers.
Microbiome dysbiosis
The epithelial barrier surfaces of our body host a diverse microbial community, or microbiota, that is composed of a variety of microorganisms, such as bacteria, fungi, and viruses[98]. Substantial studies have reported that obesity or HFD markedly affects the composition of the commercial microbiota,especially in the intestine[48]. Obesity-induced perturbation of the gut microbiota has been demonstrated to influence stem cell phenotypes. For example, structural changes in the microbiota were associated with HFD-induced myeloid progenitor skewing of the differentiation capacity of hematopoietic stem cells[99]. The intestinal tract bacteriaLactobacillusinduces the release of adiponectin by niche cells through NF-κB activation[100]. In pigs with HFD-induced obesity, the elevation of the proliferative zone and stem cell zone was associated with increased abundance of the gut bacterial phylaProteobacteriaandFirmicutes[87]. In addition to these initial data suggesting the association between obesity-related microbiome dysbiosis and the possible development of CSCs, evidence linking the microbiome in obesity to CSC function is lacking. Whether obesity-induced alterations in the composition of the host microbiota affect initiation, progression and therapy resistance and the underlying mechanisms need more investigation with the hope of providing more strategies for cancer prevention and treatment.
CLINICAL SIGNIFICANCE
If the above-discussed links between obesity and CSCs are founded on convincing evidence, an obvious question is whether targeting both can prevent or improve the outcomes of cancer. At present, targeting both obesity and CSCs has great challenges; however, progress is gradually being made. For example,bariatric surgery has been popularized globally, leading to more weight loss and longer maintenance than diet and lifestyle changes[101]. Intriguingly, in these patients with obesity who received bariatric surgery, a decrease in overall cancer diagnoses was observed[102,103]. However, surgical intervention does not guarantee the recovery of obese patients to a normal state and is typically a harmful method.Theoretically, obesity prevention represents the most promising and scientific solution, which requires joint efforts and cooperation from throughout the whole world[104]. However, under obesity pandemic conditions, exploring strategies to lower the incidence of obesity-related cancer represents the primary goal. Unfortunately, no experience has been gained. As low-grade inflammation plays a central role in linking obesity and cancer, anti-inflammatory therapy may be a promising direction, which has been validated in the prevention of colorectal cancer[105]. For obese patients with established malignancies,there is an urgent need to improve therapeutic efficacy and long-term survival. As mentioned above,various mechanisms have been proposed to link obesity and CSCs; thus, whether blocking these connections can prevent or delay the initiation and progression of cancer needs further study. Encouragingly, such strategies have been explored extensively, and some of them have already advanced into clinical use. For example, the clinical development of therapeutics targeting CSC-associated developmental signaling pathways has resulted in improved patient outcomes[82]. Some inflammatory factortargeting therapies also show promising results in improving outcomes in patients with cancer[106].Lifestyle interventions, such as reduced dietary intake and increased physical activity, were demonstrated to cause weight loss, leading to altered expression of inflammatory factors and maintenance of stem cell homeostasis[107,108]. Therefore, what awaits us next is to validate their efficacy in obese people with cancer.
CONCLUSION
As stated above, notwithstanding the clear and compelling link between obesity and CSCs, as well as an understanding of the mechanisms connecting them, this research area is still in its infancy. Most scientific research exploring the association of CSCs and obesity originates from mouse models or was inferred indirectly from the conclusions of different research fields. Therefore, there are still many major questions waiting for answers. For example, how can the profound differences in cancer incidence across different anatomical sites influenced by obesity be explained? Do the differences in the microenvironment across adult stem cell niches contribute to these variations? Regarding alterations in stem cell biology, how much overlap is there among animal models and obese patients? What role does the microbiome play in mediating the induction, maintenance and therapy response of CSCs? Does obesity differentially regulate normal and malignant stem cells, and how does it do so? How does obesity influence the crosstalk between CSCs and immunoediting? To what extent are stem cells conditioned in obesity reversed when obesity is improved? Does obesity enhance the establishment of premetastatic niches? Can strategies aimed at targeting the mechanisms linking obesity and CSCs prevent the initiation and delay the progression of cancer?
Despite a wealth of unknowns, it is clear that obesity increases the stem cell pool and induces biological modulation in these cells, which predisposes these stem cells to transformation. Regarding the increased prevalence of obesity and its convincing association with cancer, programs are urgently needed to decrease the incidence of obesity. At this time, primary obesity prevention through public health policies, including dietary and lifestyle changes, represents a compelling approach toward a reduction in the incidence of obesity and its associated cancer. Identifying obese patients with increased cancer risk and developing appropriate management of obesity or applying cancer prevention methods such as anti-inflammatory agents in these populations represents another compelling approach toward a reduction in the burden of cancer in obesity. Several mechanisms have been proposed to explain the association between obesity and CSCs, and large amounts of agents targeting these pathways have been developed. Testing them in patients with obesity and comparing their efficacy with that in nonobese individuals are important components of future translational research and clinical trials.
ACKNOWLEDGEMENTS
The author thanks the Health Commission of Mianyang City and the Science and Education Department of the Third Hospital of Mianyang for their support. The space limitations of this review have unfortunately meant that I have not been able to separately cite many of the original publications that have contributed substantially to the literature. I sincerely apologize to the authors of these publications. All figures in this review were created with BioRender.com.
FOOTNOTES
Author contributions:Li J designed the review; Li J and Xie WJ reviewed the literature; Xie WJ drafted the manuscript; Li J revised the manuscript; and all authors read and approved the final version of the manuscript.
Conflict-of-interest statement:All the authors report no relevant conflicts of interest for this article.
Open-Access:This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BYNC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is noncommercial. See: https://creativecommons.org/Licenses/by-nc/4.0/
Country/Territory of origin:China
ORCID number:Wen-Jie Xie 0000-0001-9589-9431; Jian Li 0000-0002-5807-2360.
S-Editor:Wang JJ
L-Editor:A
P-Editor:Wang JJ
 World Journal of Stem Cells2023年4期
World Journal of Stem Cells2023年4期
- World Journal of Stem Cells的其它文章
- Banking of perinatal mesenchymal stem/stromal cells for stem cellbased personalized medicine over lifetime: Matters arising
- Clinical application prospects and transformation value of dental follicle stem cells in oral and neurological diseases
- Current status and prospects of basic research and clinical application of mesenchymal stem cells in acute respiratory distress syndrome
- Extracellular vesicles: Emerged as a promising strategy for regenerative medicine
- Human pluripotent stem cell-derived B cells: Truly immature islet B cells for type 1 diabetes therapy?
- Mechanisms of analgesic effect of mesenchymal stem cells in osteoarthritis pain