
Human pluripotent stem cell-derived B cells: Truly immature islet B cells for type 1 diabetes therapy?
2023-05-25 09:24HelenJiangFangXuJiang
World Journal of Stem Cells 2023年4期
Helen Jiang, Fang-Xu Jiang
Helen Jiang, Sir Charles Gairdner Hospital, University of Western Australia, Perth 6009,Australia
Fang-Xu Jiang, School of Biomedical Sciences, University of Western Australia, Perth 6009,Australia
Fang-Xu Jiang, School of Health and Medical Sciences, Edith Cowan University, Perth 6027,Australia
Abstract A century has passed since the Nobel Prize winning discovery of insulin, which still remains the mainstay treatment for type 1 diabetes mellitus (T1DM) to this day. True to the words of its discoverer Sir Frederick Banting, “insulin is not a cure for diabetes, it is a treatment”, millions of people with T1DM are dependent on daily insulin medications for life. Clinical donor islet transplantation has proven that T1DM is curable, however due to profound shortages of donor islets,it is not a mainstream treatment option for T1DM. Human pluripotent stem cell derived insulin-secreting cells, pervasively known as stem cell-derived B cells (SCB cells), are a promising alternative source and have the potential to become a T1DM treatment through cell replacement therapy. Here we briefly review how islet B cells develop and mature in vivo and several types of reported SC-B cells produced using different ex vivo protocols in the last decade. Although some markers of maturation were expressed and glucose stimulated insulin secretion was shown, the SC-B cells have not been directly compared to their in vivo counterparts, generally have limited glucose response, and are not yet fully matured. Due to the presence of extra-pancreatic insulin-expressing cells, and ethical and technological issues, further clarification of the true nature of these SCB cells is required.
Key Words: Human pluripotent stem cells; Stem cell-derived B cells; Islet B cells; Type 1 diabetes mellitus; Cell replacement therapy
INTRODUCTION
In this coronavirus disease 2019 pandemic era, there is a silent growing epidemic of significant public health burden with tremendous social and economic costs. This growing epidemic is not an infectious disease, but a chronic non-communicating metabolic disease - it is the epidemic of diabetes mellitus(DM). There was an estimated 537 million adults with DM globally in 2021[1], with the prevalence increasing each year due to the rising incidence of type 2 DM (T2DM) worldwide[2]. DM is a metabolic disorder characterised by a disruption in glucose homeostasis leading to hyperglycaemia, and broadly consists of 2 main types: T1DM and T2DM. T1DM is the absolute deficiency of insulin due to the autoimmune destruction of insulin-secreting B cells in the islets of Langerhans of the pancreas, and is usually first diagnosed in children and young adults. T2DM is the relative deficiency of insulin function due to insulin resistance in peripheral tissues, and sometimes with reduced insulin secretion due to dysfunctional or dedifferentiated B cells, usually occurring in adults.
Hyperglycaemia in T1DM and T2DM can cause various microvascular complications such as diabetic retinopathy and blindness, nephropathy and kidney failure requiring dialysis, as well as peripheral neuropathy and infected foot ulcers that lead to amputations. It can also cause macrovascular complications such as peripheral artery disease, coronary artery disease and stroke. These complications lead to significant morbidity and mortality, as well as substantial associated health and social costs[3,4].
INSULIN IS NOT A CURE FOR DIABETES
These were Sir Frederick Banting’s words to the world during his Nobel Lecture for his 1923 Nobel Prize winning discovery of insulin. Subsequent discoveries on primary insulin sequences and radioimmune assay for insulin and other peptide hormones were also awarded the Nobel Prizes(Figure 1)[5]. A century later, unfortunately there is still no cure for DM, and life-long insulin replacement remains the mainstay of treatment for T1DM and controlling high blood sugar levels with antihyperglycaemic agents in most T2DM individuals. The continuous blood glucose monitoring and insulin pump known as artificial pancreas or bionic pancreas still presents the risk of developing complications, though reduced, because this and other current treatments cannot achieve physiological glucose homeostasis in patients[6,7]. These treatments themselves are also not without risks. Insulin as well as some oral anti-hyperglycaemics, such as sulfonylureas and glinides, are associated with the risk of hypoglycaemia which can lead to seizures, coma and even death[8,9]. Thus, there is a critical need for more effective and curative treatments to reduce the global burden of this disease.
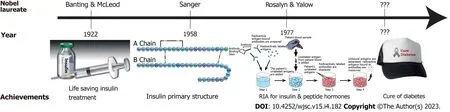
Figure 1 Nobel prizes awarded for the endeavour towards curing diabetes.
The landmark proof-of-concept has demonstrated over the last 2 decades that clinical transplantation of donated human islets are able to restore B-cell function and achieve insulin independence immediately with improvement in glycaemic control and avoid the risk of hypoglycaemia episodes[10-13]. However, a large amount of approximately 340-750 million islet cells are required for successful transplantation in a patient of 68 kg weight[11,14]. Thus, the widespread application of donor islet transplantation is severely limited by the insufficient supply of human organ donor pancreases[15,16].In addition to supply issues, another challenge to this treatment option is the prevention of transplant rejection, immune destruction and cell death of the transplanted islet cells[17]. To address the donor shortage issue, alternative scalable insulin-secreting tissues must be identified and developed. Due to their ability for theoretically infinite self-renewal and differentiation into all cell types in the body,human pluripotent stem cells (hPSCs) hold great promise for generating surrogate insulin-secreting cellsex vivo, pervasively known as stem cell-derived B cells (SC-B cells)[18-20] or SC-islets[21] in the literature. In order to help understand the true nature of these SC-B cells, we briefly introduce how islet B cells developin vivo.
IN VIVO DEVELOPMENT OF ISLET BETA CELLS
The pancreas is derived from the embryonic endoderm, one of the three germ layers, which is formed during gastrulation of embryogenesis. In addition to the pancreas, the definitive endoderm also gives rise to the liver, lung, thymus and other organs of the respiratory and digestive tracts[22]. The endoderm located in the foregut region gives rise to the dorsal and ventral buds of the pancreas which rotate to form one organ, then pancreatic epithelium is induced and expands, from which endocrine progenitors arise. The endocrine progenitors then differentiate into the B cells that secrete the hormone insulin, α cells that secrete the hormone glucagon, δ cells that secrete the hormone somatostatin, ε cells that secrete the hormone ghrelin, and PP cells that secrete the hormone pancreatic polypeptide. The pancreatic endocrine cells start to organize into clusters forming islets before birth, and the Islets of Langerhans become fully formed at around 2-3 wk after birth[22]. Human islets are made up of 40%-60% B cells and 30% α cells[22]. The adult pancreas is made up of exocrine cells that secrete digestive tract enzymes, duct cells that make up the ductal tree to transport digestive enzymes and islet cells that secrete hormones into the bloodstream for glucose homeostasis[23].
Mechanistically, the pancreatic islets are initiated by the transient expression of a high level of the transcription factor neurogenin-3 (NGN3)[24]. NGN3 is important in committing all pancreatic endocrine cell types, the deficiency of which leads to the absence of pancreatic endocrine cells[25,26].The molecular mechanisms for the development of each pancreatic endocrine cells are not completely defined, however it is suggested that insulin-producing B cells are differentiated from the pancreatic progenitors that express transcription factor genes pancreatic and duodenal homeobox 1 (PDX1) and NK6 homeobox 1 (NKX6-1),and then turn on NGN3[27,28]. There are several B cell transcription factors, including PDX1, NKX6-1 and MAF BZIP transcription factor A (MAFA), which play a critical role in activating insulin transcription and regulating insulin secretion[29-31].
PDX1 is a homeodomain transcription factor homogenously expressed in the early pancreatic bud and its expression persists into mature B cells; the absence of PDX1 leads to agenesis of the pancreas[32]. NKX6-1 and the helix-loop-helix transcription factor Beta2/NeuroD determine islet cell differentiation during embryogenesis, and maintain specific islet cell hormone expression in adults[32].Knockout of mouseNkx6-1gene leads to a significant inhibition in the formation of B cells[33]. NeuroD is initially expressed in pancreatic epithelium during development, before being expressed in NGN3+endocrine progenitors, and finally exclusively expressed in B cells after birth. The absence of Beta2/NeuroD leads to reduced mouse endocrine cells, in particular B cells, increased apoptosis and arrestment in islet morphology[25,34]. Beta2/NeuroD is also a critical transcriptional activator of the insulin gene[35,36].
B-cell maturation including maturation of other clinically important cell types is a postnatal development process. For example maturation of mouse and human B cells takes place approximately 3 wk[37] and 26-44 wk after birth[38,39] respectively. The maturation process is controlled by transcription factors and exhibited in maturing at the gene, protein, subcellular, intercellular and metabolic levels.
ISLET BETA CELL MATURATION REGULATED BY TRANSCRIPTION FACTORS
Following B-cell specific NeuroD deletion, the mice developed glucose intolerance and the islets displayed features of foetal/neonatal B cells such as overexpression of glycolytic genes, lactate dehydrogenase (LDHA), Neuropeptide Y, and higher basal insulin secretion and oxygen consumption due to the reliance on oxidative metabolism of glucose[40-46]. That is, the glucose metabolic profile of mouse B cells without NeuroD was equivalent to immature B cells. The mutations of NeuroD cause maturity onset diabetes of the young[22,33]. NeuroD is also critical for maintaining a matured functional state of islet B cells[40]. These data suggest that NeuroD regulates islet B-cell maturation, though its postnatal dynamic expression profile is not available. Thus, identification of Beta2/NeuroD activators may help mature hPSC-derived insulin-secreting cellsex vivo.
MafA is another transcription factor being demonstrated to regulate the maturation of islet cell organisation, B cell mass and B cell function from 3 wk of age in mice using the gene targeting strategy[47]. MafA expression reaches their adult levels at 3 mo in rats[48] coinciding with the obtaining of mature glucose stimulated insulin secretion (GSIS). Aguayo-Mazzucatoet al[49] were the first to demonstrate that MAFA overexpression and the thyroid hormone triiodothyronine (T3) treatment are able to increase human foetal islet-like clusters, insulin secretion at 16.8 mmol/L glucose and proinsulin-to-insulin processing. Chromatin immunoprecipitation experiment showed binding of thyroid receptors to MafA promoter, thereby confirming that T3 directly regulates the expression of MafA[50]. The thyroid hormone receptor is also demonstrated to be expressed on human mature islets[51], though its postnatal development profile is unknown.
Furthermore, a recent study shows that the expression of the orphan nuclear transcription factor estrogen-related receptor gamma (ERRγ) is a hallmark of mature B cells[52]. ERRs consist of three paralogs in mammals, namely ERRα (NR3B1 or Esrrα), ERRB (NR3B2 or EssrB) and ERRγ (NR3B3 or Essrγ). ERRγ is progressively upregulated in mouse islets from 2 to 6 wk of age (5-fold higher in adults compared to neonatal B cells) and ERRγ transcriptional network promotes mitochondrial oxidative metabolism in mouse B cells, required for functional maturation of B cells and glucose homeostasis[52].Mice with B cell-specific ERRγ deletion failed to develop a mature GSIS. With the developmentally deleted B cell-specific ERRγ knockout mouse islets, RNA sequencing (RNA-seq) revealed that the expression of 4189 genes were altered, with almost equal numbers of genes down- and up-regulated(2008 and 2182 genes respectively). Gene ontology analysis revealed that ERRγ-regulated genes are associated with processes critical for B cell function including ATP biosynthesis, cation transport,oxidative phosphorylation, electron transport and secretion[52]. However, data is not available on postnatal developmental expression of ERRs in human islet cells, which will have to be addressed in the near future. Identification of ERRγ activators may help mature hPSC-derived insulin-secreting cellsex vivo.
Expression of theSine Oculisfamily of homeodomain transcription factors SIX2 and SIX3 increased with age in the human pancreatic islet B cells[53]. SIX2 and SIX3 are localised to the nucleus of adult human B cells but not detected in juvenile (under 9 years of age) B cells[53]. Using gain-of-function experiments in human B cell line, the EndoC-bH1 cells or primary juvenile human islets, evidence has demonstrated that expression of SIX2 or SIX3 were sufficient to enhance cardinal functions of human B cells[53]. Identification of SIX2 and SIX3 activators may therefore help generate matured hPSC-derived insulin-secreting cellsex vivo.
METABOLIC MATURATION OF ISLET BETA CELLS
Although rat islets acquired GSIS by postnatal day 21, a mature GSIS was only achieved by 3 mo[54],coinciding with the time when insulin dynamics reaches their adult levels[48]. The metabolic maturation is underscored by genes of important metabolic players in B cells such as glucose transporter 2, glucokinase, glucagon-like peptide-1 receptor and prehormone convertase 1 (encoded byPcsk1) that were expressed from very low levels at postnatal day 2 to higher levels with increased age[48].Similarly, the metabolic maturation is also underscored by genes transcribing for malate dehydrogenase, glycerol-3-phosphate dehydrogenase, glutamate oxaloacetate transaminase, pyruvate carboxylase and carnitine palmitoyl transferase 2 from much lower levels at neonatal postnatal day 2 to high levels at day 28[55]. In the same period, genes encoding proliferation regulators in B cell genes encoding platelet-derived growth factor receptor A, platelet-derived growth factor receptor B, plateletderived growth factor B and fibroblast growth factor (FGF) receptor 1 are progressively downregulated.Mature B cells tend to have lower levels of LDHA and glycolytic genes, as it is downregulated by NeuroD from embryonic to adult islets, which then appears to possess the ability to correspond glucose metabolism with insulin secretion[56-58].
The molecular mechanism of mature B-cell secretion is well understood. The higher blood glucose levels stimulate its active transportation into the B cell cytoplasm, increasing the ATP/ADP ratio through glycolysis and oxidative metabolism and triggering the depolarisation of the B cell membrane and opening the voltage-gated Ca2+channel. The AMP-activated protein kinase (AMPK) is a highly conserved sensor of intracellular adenosine nucleotide levels that is activated even with modest decreases in ATP production resulting in relative increases in AMP or ADP. In response, AMPK promotes catabolic pathways to generate more ATP, and inhibits anabolic pathways. The increase in cytoplasmic Ca2+triggers the fusion of insulin granules with the plasma membrane for exocytosis of insulin contents. Coordinating with other islet cells, mature B cells produce insulin in adequate amounts and timeliness to maintain plasma glucose within a narrow concentration range[39,59]. Thus, B cell function is critical for mature GSIS. Destruction and failure of islet B cells will lead to T1DM and T2DM,respectively (Figure 2).
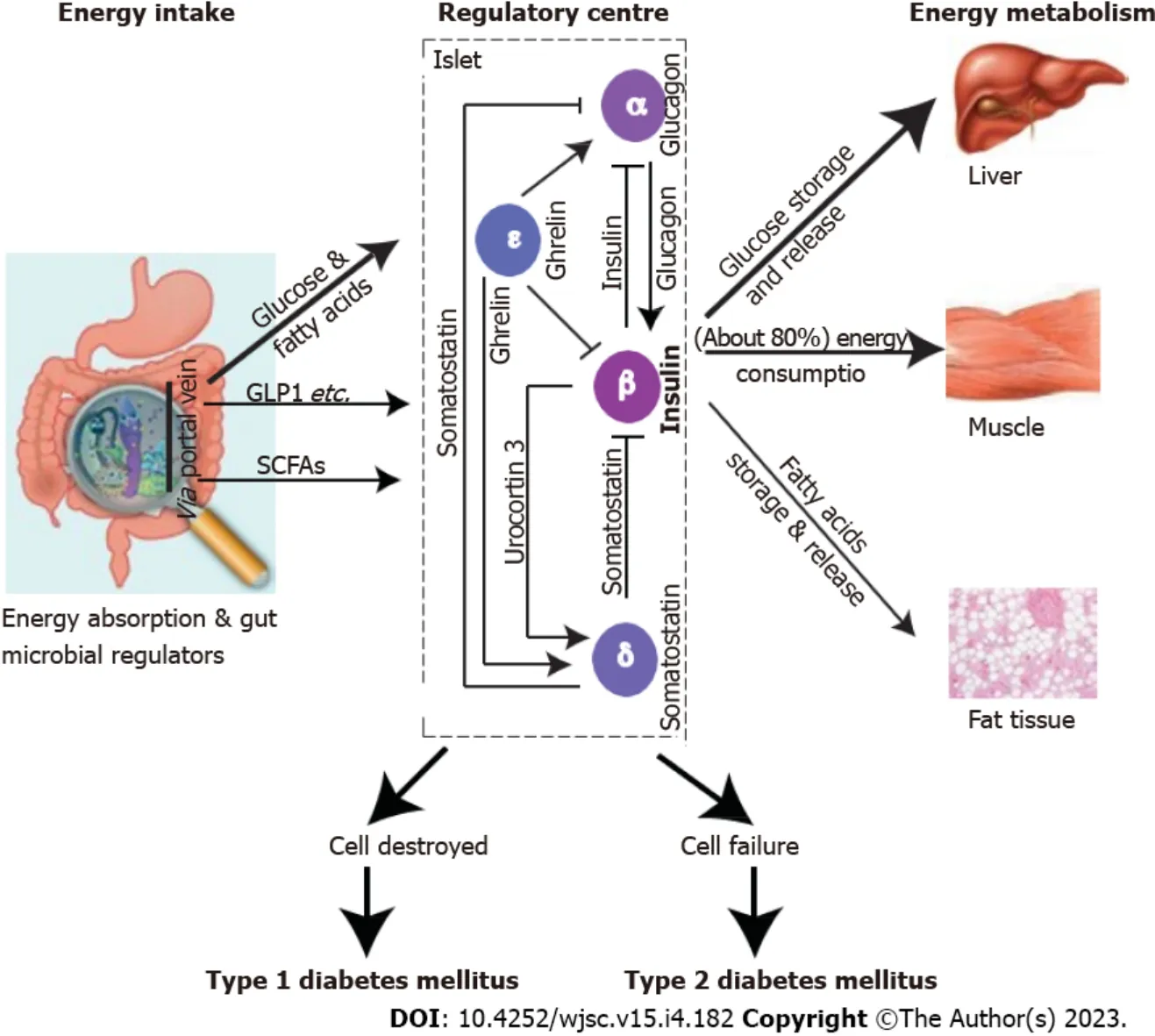
Figure 2 Mature islets are a regulatory centre for glucose homeostasis.
MATURATION MARKERS OF ISLET BETA CELLS
To help with the characterization of whether hPSC-derived insulin-secreting cellsex vivoare matured,we briefly summarize maturation markers forin vivoislet B cells. Over the last decade, several potential markers for maturation of immature islet B cells were discovered. Blumet al[60] were the first to demonstrate that functional islet B cell maturation is marked by expression of the corticotropin-releasing factor family peptide urocortin 3 (UCN3), along with an increased glucose threshold.
Mature rat B cells expressed significantly higher levels of the gap junction connexion 36 gene (Cx36,also known asGjd2) compared to neonatal immature counterparts, corresponding to a significantly higher membrane density of gap junctions and greater intercellular exchange of ethidium bromide[61].Human mature islets predominantly express CX36 at mRNA and protein levels with B cell membrane harboring detectable levels of CX36 gap junction proteins[62]. Though the developmental profile of human islet CX36 is unknown, we speculate that the dynamic pattern of CX36 expression from human neonatal to mature B cells is similar to that in rats and CX36 is a potential maturation marker for matured hPSC-derived insulin-secreting cellsex vivo.
Our group recently showed that claudin 4 is the only tight junction molecule family member highly upregulated in the postnatal mouse islets and global deletion of this gene affects mature GSIS in a sex difference manner[63]. Thus, claudin 4 may also be a maturation marker for matured hPSC-derived insulin-secreting cellsex vivo.
THE DIFFERENTIATION OF SC-BETA CELLS EX VIVO
The advent of hPSC provided an important opportunity to overcome major challenges of clinical islet transplantation therapy through its accessibility, theoretically unlimited self-renewability and the boundless potential to generate an alternative source of donor insulin-secreting cellsex vivo[64,65]. The generated insulin-secreting cells can also be used for disease modelling and pharmaceutical drug testing to help establish therapeutics that improve cell function, survival and proliferation. Insulin-secreting cells differentiated from hPSCs that include human embryonic stem cells (hESC) and induced hPSCs(ihPSC) are ubiquitously termed as SC-B cells[18-20] in the literature. hESCs are generated from the inner cell mass of human blastocysts and have the infinite ability to proliferate as undifferentiated cells or differentiate into cells of all ectoderm, mesoderm or endoderm lineages[66,67].
Over recent years, there have been various protocols developed ofex vivodifferentiation of SC-B cells[18-20] and SC-islets[18,21]. Thus far, hPSCs have been differentiated towards SC-B cells through a stepwise manner emulatingin vivopancreatic embryonic development[68-72]. The differentiation of hPSCs towards SC-B cells have been achieved with the application of growth factors, proteins or molecules to modulate signaling pathways to progress through each stage of pancreatic development, and is usually measured by expression of a couple of key transcription factors or C-peptide[22]. hPSCs (characterised by expression of Oct4) are first differentiated into definite endoderm cells expressing FOXA2 and SOX17 through application of a mix containing Wnt, activin A, inducer of definite endoderm, wortmannin, and sodium butyrate[22]. Then application of FGF10 and FGF7 differentiates the definite endoderm into gut tube endoderm expressing HNF1B and HNF4A[22]. The differentiation mixture containing retinoic acid,noggin KAAD-cyclopamine, FGF, and indolactum V leads to differentiation into pancreatic progenitors expressing PDX1 and HNF6, which further differentiates into endocrine progenitors (NKX6-1, NGN3,NKX2-2, PTF1A), and finally into B-cells (characterised by presence of C-peptide and insulin)[22]. We here summarize several representative protocols used to generate SC-B cells and SC-islets (Table 1).
Pagliucaet al[73]’s differentiation protocol was the first using specific and cocktail of inducing factors to differentiate hPSCs sequentially through 6 stages into SC-B cells (Table 1). At stages 5 and 6, there is however significant heterogeneity in the final population containing SC-B and SC-α cells, as well as SCendocrine cells (resembling enterochromaffin cells) and non-endocrine cells (e.g.,exocrine cells such as pancreatic acinar, mesenchymal and ductal cells)[18]. Nevertheless, these cells appear to be stable,maintaining their identity as evidenced by their global transcriptional profiles during stage 6 cultures.At this stage, they also express the maturation marker SIX2 but several other B cell markers of maturity are not expressed such as UCN3, MAFA and SIX3[18].
The Velazco-Cruzet al[19]’s protocol was built upon and modified Pagliucaet al[73]’s protocol, and demonstrated that the SC-B cells had improved insulin secretion and greater gene expression of B cell markers compared to the cells generated with Pagliucaet al[73]’s protocol, but still much less than the average human islet (Table 1). Follow-up studies with the addition of differentiation factors or changes to the differentiation processes were unfortunately unsuccessful in producing more functional SC-B cells equal to human islet B cells[74-76].
In Balboaet al[20]’s protocol, the SC-islets had similar cytoarchitecture and functional insulin secretion pattern to islet B cells, though with immature glucose-induced mitochondrial respiration and instead retained pyruvate sensitivity - thus the SC-islets were not completely similar to functional adult islets (Table 1). Balboaet al[20]’s SC-B cells showed heterogeneous mature B cell marker expression,required further maturationin vivoafter transplantation, showed upregulated expression of CHGB and MAFA after 6 mo, and did not express adult B cell factors RBP4 and SIX3[20].
Nevertheless, studies indicate that several current pancreatic progenitor differentiation protocols promote precocious endocrine commitment; ultimately resulting in the generation of non-functional polyhormonal cells[74]. The efficiency of differentiation decreases with each step, and at the final step there are very small amounts of SC-B cells that have a low insulin content, co-express insulin and glucagon, and usually respond poorly to glucose stimulation[22,70]. It was also found that these SC-B cells have little to no expression of maturation genes including MAFA and G6PC2[18,73,77]. Following transplantation, the amount of insulin secreted by SC-B cells rises[73,77,78] and the previously low or non-expressed genes of islet B cells such as MAFA, G6PC2, MNX1 and INS increases[79].
Cell purification steps will increase the safety of, and ability to upscale the manufacture of B cells.However, there are difficulties in including this step in large-scale manufacturing processes for production of reproducible PSC-derived cellular products with less variability in composition and function[80]. Several cell surface markers have been used to purify different developmental stages of PSC-derived cells[80]. Markers used include CD177 for anterior definitive endoderm cells[81], CD142,CD24 and glycoprotein 2 for pancreatic progenitors[82-85], CD49a for SC-B cells[18], and CD9 for negative selection of SC-B cells[86]. Monoclonal antibody against extracellular domain of claudin 4 might help enrich matured SC-B cells differentiatedex vivo.
Finally, a few maturation factors have proven useful in maturing SC-B cellsex vivo. For example, T3 enhanced the MAFA expression in the SC-B cells, and increased insulin content and insulin secretion at 16.8 mmol/L glucose[49]. Using an adenoviral ERRγ vector, overexpression of ERRγ increased glucosestimulated C-peptide secretion in hPSC-derived insulin-secreting cells, thus may promote their functional maturation[52]. Therefore, identification of molecules that activate NEUROD, ERRγ, SIX2 and SIX3 will be important. Application of the activators individually or in combination may indeed promote functional maturation of genuine SC-B cells.
CURRENT EX VIVO SYSTEMS ARE DISTINCT FROM IN VIVO ISLET DEVELOPMENT NICHES
The current PSC differentiation protocols for insulin-secreting cells are mostly bulk cultures and consist of cocktails of inducing factors, which are generally based on accumulative knowledge generated from using the animal model systems. In these bulk cultures, there are cells types in the targeted lineage aswell as unwanted lineages. One or twoin vivobiomarkers are selected based onin vivoislet lineage development to characterise targeted cells at different differentiated stagesex vivo. However, thesein vivobiomarkers should not be extrapolated as biomarkers for theex vivodifferentiation conditions because of clear differences in spatiotemporal and microenvironment niches between thein vivodevelopment andex vivodifferentiation (Figure 3). In other words, we do not yet fully understand the full regulatory program, or the molecular details of the 3D microenvironment niche for specific islet lineage developmentin vivoto guide the specific differentiation of hPSCs into insulin-secreting cellsex vivo.
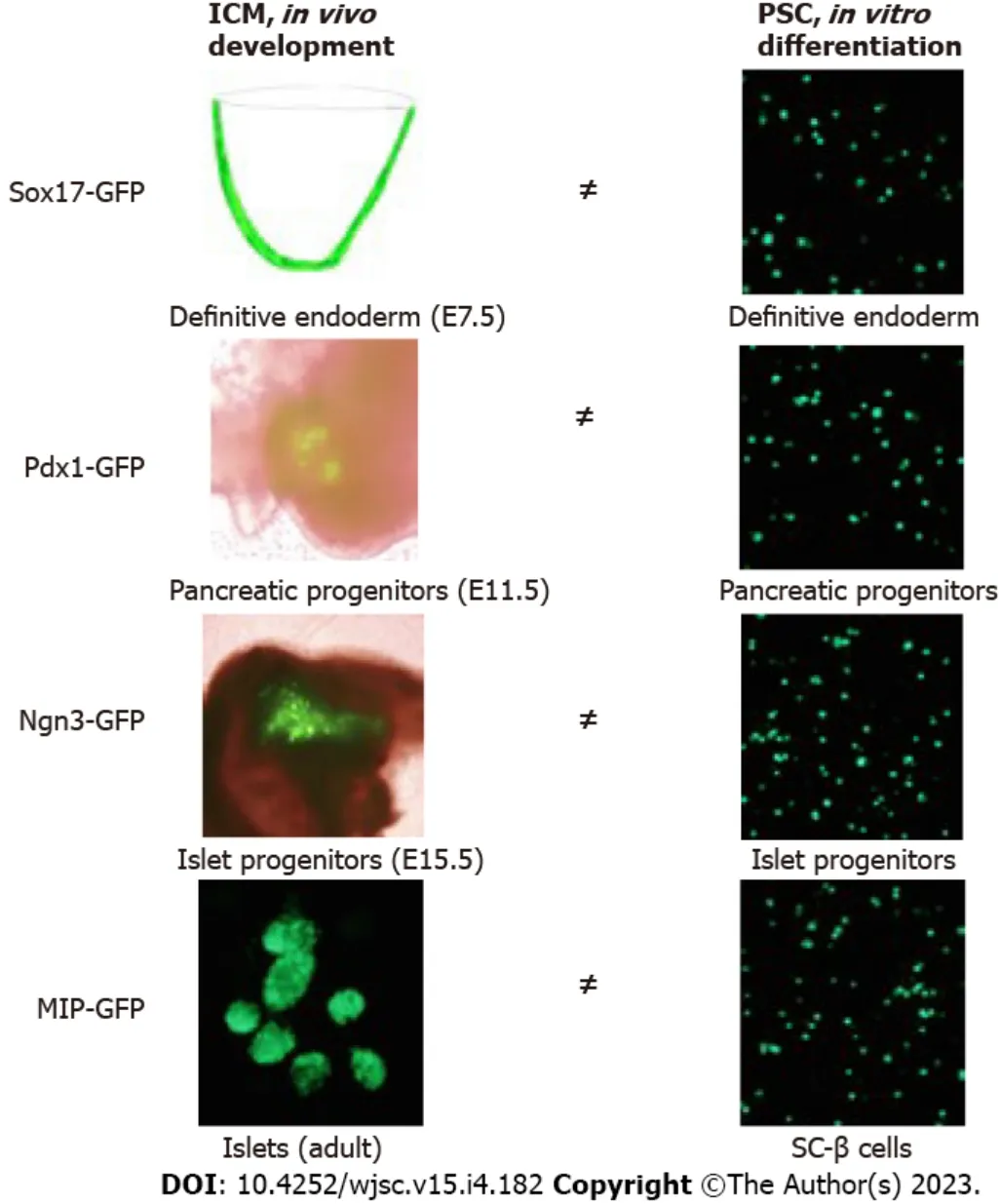
Figure 3 There are clear differences in spatiotemporal and microenvironment niches between the in vivo development and ex vivo differentiation of islet lineages.
EXTRAPANCREAS INSULIN-SECRETING CELLS
Perhaps the research community have also forgotten the fact that in our body, extra-pancreas insulinsecreting cells exist, which may complicate the efforts of generating genuine SC-B cells. Subverted to general knowledge, approximately a quarter of human foetal enteroendocrine K/L cells were recently shown to express high levels of insulin and other B cell genes including the transcription factor PDX1,by using samples of foetal and neonatal human small intestines derived from the endoderm during development[87]. Notably, the expression of UCN3 in the human foetal enteroendocrine K/L cells was higher than in foetal human pancreatic B cells[87]. These results were confirmed with single molecule fluorescencein-situhybridisation of insulin mRNA combined with immunofluorescent antibody staining of the insulin protein[87]. Secondly, thymocytes that are derived from the foregut, adjacent to which gives rise to the pancreas, normally produce insulin to induce self-tolerance and protect the body from the autoimmune destruction of pancreatic insulin-secreting B cells[88]. Lastly, though the central nervous system is an ectoderm-derived organ, the neuronal progenitors derived from adult hippocampus and the olfactory bulb were demonstrated to undergo insulin biosynthesis[89]. HumanINSmRNA expression is also detected in the hippocampus, amygdala and temporal lobe in addition to the olfactory bulb, cerebellar and pontine regions[90]. A historical account of the extrapancreas insulinsecreting cells is referred to in a recent review article[88]. These data suggest that it is possible that the current reported SC-B cells contain a varied percentage of non-pancreatic insulin-secreting cells. Future studies are required to increase the percentage of genuine insulin-secreting B-like cells in theex vivosystems.
SC-BETA CELLS TRANSPLANTED INTO NON-HUMAN PRIMATES
To further test their functions, the chemically induced SC-islets were recently intraportally transplanted into immunosuppressed adult diabetic rhesus macaques[21]. Three months after the SC-islet transplantation, all four macaques reportedly had improvements in diabetic symptoms, glycaemic control, fasting blood sugar levels, hemoglobin A1c (HbA1c), and reduced exogenous insulin requirements[21]. However, after 5-6 mo, two of the macaques developed graft failure (the other two macaques died of immunosuppression-related complications)[21]. Autopsy conducted on the macaques found no evidence of teratoma or tumorigenesis, but levels of B cells had fallen. The authors concluded that the immunosuppression regimen used was not appropriate in preventing immune attack against the grafts[21]. Whether the short-term improvements in diabetic rhesus macaques are related to the immaturity of grafted SC-islets and/or the presence of non-pancreatic insulin-secreting cells needs to be determined in the future.
SC-BETA CELLS IN CLINICAL TRIALS AS A T1DM THERAPY
The first hPSC-derived, differentiated cell replacement T1DM therapy product named VX-880 was approved by the United States Food and Drug Administration for phase 1/2 clinical trials in March 2021. The VX-880 are SC-islets for T1DM patients with certain indications; that is, impaired hypoglycaemic awareness and severe hypoglycaemia[91]. The preliminary outcomes of the clinical trials were presented in June 2022 at the American Diabetes Association 82ndScientific Sessions by Vertex, a United States Pharmaceutics company[91]. A half-dose of VX-880 in two patients was able to achieve glucose-responsive insulin secretion, significantly improve time-in-range (the amount of time that blood glucose level is measured to be within target blood sugar range 70-180 mg/dL or 3.9-10 mmol/L),reduce exogenous insulin requirements and improved HbA1c[91]. VX-880 was also well tolerated although with some largely mild or moderate adverse reactions[91]. For example, patient 1 showed blood glucose time-in-range change from 40.1% on 34.0 units per day of exogenous insulin at baseline to 99.9% and insulin independence at day 270 onwards. Patient 2 showed blood glucose time-in-range change from 35.9% on 25.9 units per day of exogenous insulin at baseline to 51.9% with a 30% reduction in exogenous insulin use at day 150[91]. Whereas these results are very promising, VX-880 requires a lengthyin vivomaturation period for blood sugar control (in patient 1) in contrast to donated islets retrieved from deceased persons, which achieved immediate insulin independence after transplantation into recipients[10-13]. The lengthyin vivomaturation period of grafted VX880 is a strong independent indicator that these SC-B cells are immature. It is also premature to claim the VX-880 SC-B cells are all genuine immature counterparts of islet B cells, as the duration and longevity of insulin independence was not yet available at the time of writing this article.
CONCLUSION
Immaturity of PSC-derived cells is a general obstacle, not only in the case of SC-B cells and SC-islets, but also other clinically important cell types[92]. Maturation biology is the final frontier in stem cell biology,of which our knowledge is still in its infancy. As summarised in Table 1, multiple hPSC differentiation protocols have been used in different laboratories. Consequently, off-target differentiation and aberrant differentiation from these protocols are more likely unavoidable, resulting in only a low frequency of genuine SC-B cells. Furthermore, the stage-specific differentiation factors selected may direct nonspecific spatiotemporal differentiation, thus resulting in multiple cell types of the endodermal germ layer and even neuronal lineage origins. This may result in some differentiating cells along unwanted pathways and give rise to extrapancreas insulin-secreting cells. On the other hand, unwanted or offtarget differentiated cellular products have accumulated in the bulk culture protocols and not been excluded for subsequent differentiation steps, which further increases the possibility of compromising the characterization through use of one or two developmental markers ofin vivocellular lineages.Finally, in addition to the above, there are still other challenges in this exciting field of research, such as ensuring SC-B cell survival post-transplantation given the highly vascularised islets are susceptible to ischaemic injury and loss of cell mass[93,94]. Developing methods that evade autoimmune attack in T1DM patients without the use of lifelong immunosuppression would be valuable[95].
Stage-specificin vivopancreatic and islet lineage cell types would provide ideal positive controls for theex vivohPSC-derived insulin-secreting cells. Nevertheless, the human ethics issues and lack of human embryonic and foetal pancreatic tissues available prevent such reliable and precise comparison to be made between the islet lineage cells and the PSC-derived cells. However, future efforts should be made to resolve these issues. Similarly, it would be wise not to solely concentrate on undertaking human B cell differentiation and maturation studies from hPSCs. Instead, investigating B cell differentiation and maturation from model animals will be invaluable and will facilitate the realisation of a curative stem cell therapy for people with T1DM.
In order to minimise confusion between theex vivodifferentiated insulin-secreting cells and islet B cells, our laboratory proposed a 4-criterial post-genomic concept for naming “B cells” a few years ago[96]. Recently, Kaestneret al[97] described many islet biologists/scientists much like the “Parable of the Blind Men and the Elephant” in terming “B cells”. This appears to be the case in respect to claims made about SC-B cells without proper positive controls of correspondingin vivoislet lineage cells. Kaestneret al[97] further proposed six salient features of normal, fully functional mature B cells and made a recommendation to not name PSC-derived insulin-producing cells as “B cells”, but conservatively as insulin-producing cells, insulin+cells or B-like cells, when there is no clear evidence that the six features ofin vivoB cells are met.
The degree of single-cell RNA-seq (scRNA-seq) data similarity between the SC-B cells and donated islet B cells remains largely unclear. First, all scRNA-seq datasets of SC-B cells lacked a direct positive control fromin vivopancreatic and islet lineage cells. Second, most current scRNA-seq methods provide a high throughput but sacrifice full transcript coverage and sensitivity[98]. Third, as barcodes/inducers are introduced by the template switching of reverse transcriptase, strand invasion becomes problematic through systematic bias, namely biases from the introduction of artefacts. Fourth, loss of cDNA synthesis and bias in cDNA amplification leads to severe quantitative errors of these scRNA-seq methods[99]. Fifth, the current scRNA-seq methods suffer from impaired mRNA accounting. However,molecular spikes have significantly improved single cell mRNA accounting[100], adoption of the molecular spike method and further improvements may help address the above issues. As such,genuine SC-B cells will eventually become available as donor cells for establishing curative therapies for people suffering from T1DM in the not too distant future.
FOOTNOTES
Author contributions:Jiang H wrote the first draft of the manuscript; Jiang FX conceived and designed the research;and all authors have edited, revised, reviewed and approved submission of the manuscript.
Supported bythe Juvenile Diabetes Research Foundation, No. 4-2006-1025; Diabetes Australia Research Trust; and Telethon Perth Children’s Hospital Research Fund (TPCHRF) grant to Jiang FX.
Conflict-of-interest statement:All the authors report no relevant conflicts of interest for this article.
Open-Access:This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BYNC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is noncommercial. See: https://creativecommons.org/Licenses/by-nc/4.0/
Country/Territory of origin:Australia
ORCID number:Helen Jiang 0000-0002-5949-141X; Fang-Xu Jiang 0000-0003-3172-0476.
S-Editor:Wang JJ
L-Editor:A
P-Editor:Wang JJ
 World Journal of Stem Cells2023年4期
World Journal of Stem Cells2023年4期
- World Journal of Stem Cells的其它文章
- Banking of perinatal mesenchymal stem/stromal cells for stem cellbased personalized medicine over lifetime: Matters arising
- Obesity and cancer stem cells: Roles in cancer initiation,progression and therapy resistance
- Clinical application prospects and transformation value of dental follicle stem cells in oral and neurological diseases
- Current status and prospects of basic research and clinical application of mesenchymal stem cells in acute respiratory distress syndrome
- Extracellular vesicles: Emerged as a promising strategy for regenerative medicine
- Mechanisms of analgesic effect of mesenchymal stem cells in osteoarthritis pain