
钛基体Ti4O7阳极在高盐体系下的电催化降解性能
2023-05-05 03:13:50郭思远王雪李昕圆荆晓生龙志徐浩延卫贾亮
西安交通大学学报 2023年4期
郭思远,王雪,李昕圆,荆晓生,龙志,徐浩,3,延卫,3,贾亮
(1. 西安交通大学环境科学与工程系,710049,西安; 2. 河南龙兴钛业股份有限公司,454650,河南济源; 3. 浙江西安交通大学研究院,311200,杭州)
电催化氧化是一种操作便捷、绿色安全、能耗低、二次污染小、节约占地面积的电化学水处理技术[1-2]。作为电催化氧化技术的核心,阳极的选择至关重要[3]。目前的商用电极种类中:传统的金属电极和钛基体金属氧化物电极(也称为形隐电极(dimension stable anode,DSA),包括PbO2电极[4-5]、Sb-SnO2电极[6]和Ir/Ru/Ta系列电极)均存在表面金属元素溶解现象,会造成一定的安全隐患;碳素电极的催化性能波动较大,Sb-SnO2电极和Ir/Ru/Ta类电极表面的羟基自由基产量不高,催化能力有待改善;贵金属电极和掺硼金刚石电极的制备成本较高,DSA电极制备过程中也需使用贵金属盐类配制刷涂液,经济方面不利于规模化使用[7]。此外,DSA电极在使用过程中(尤其是有F-或高浓度Cl-的条件下),均存在表面活性元素溶解、表面氧化物层溶蚀脱落以及钛基体钝化等现象,由此导致电极发生不可逆失活,实用稳定性需要进一步改进提升[8]。所以,亟需寻找新的电极材料开发新的电极材料以满足电催化氧化技术的需求。
近几年,亚氧化钛材料成为研究热点,其在金属-绝缘体过渡、燃料电池和污水处理等方面的应用均有文献报道[9]。研究表明,亚氧化钛材料具有高导电性、优越的化学稳定性、宽电化学稳定电位窗口等独特的物理和电化学性质[10],满足了电催化氧化体系中选择电极的需求。其中,Ti4O7材料的电导率最优,可以达到1 500 S/cm,约为石墨电导率(727 S/cm)的2倍[11]。Wang等[12]通过对比3种类型的亚氧化钛阳极与其他典型的活性和非活性阳极对对硝基苯酚(PNP)溶液的电氧化降解效果,证实了以Ti4O7为主体的Magnéli相阳极的优越性。与上述材料相比,Ti4O7电极在安全性、催化性、经济性、稳定性等方面均可以满足日益复杂的工业有机废水处理需求[9]。
高盐有机废水是指总含盐量(质量分数)大于1%的废水,主要来源为石油化工、煤化工、医药、印染等生产过程。它作为一种难降解有毒有机污染物废水,存在来源广泛、含盐量大、成分复杂、可生化性差等处理难题[13]。目前,常用的处理方法主要有生物法和物化法。但是,由于生物法需培养特定微生物,处理周期较长。物化法运行成本较高、处理容量较小。所以,这两种处理方法的应用受到限制[14]。电催化氧化技术不仅可以有效解决上述难题,而且可以利用溶液中的氯离子原位产生具有强氧化性的次氯酸加速有机污染物的降解,使得降解反应从电极附近的区域扩展到整个体系,扩大反应位点,加速降解[15],变废为宝。但是,处理此类废水对电极材料的机械强度和稳定性方面有极高要求。文献[16]对比了Ti/Ta2O5-IrO2、石墨及Ti/RuO2-IrO2这3类电极处理高盐废水的效果,结果表明:DSA电极优于石墨电极,但处理后的废水虽仍含有一定含量的化学需氧量(COD);采用等离子喷涂技术制备的Ti4O7电极涂层致密,粘结强度高[12],理论上可以在高盐体系下对污染物进行有效电催化氧化降解。
本文采用等离子喷涂技术制备Ti4O7电极,通过扫描电子显微镜(SEM)、X射线衍射(XRD)、X射线光电子能谱(XPS)、循环伏安扫描(CV)、电化学阻抗测试(EIS)和强化寿命测试等对电极性能进行表征并分析电极失效机制,然后以制得电极作为阳极在高盐体系下对酸性红G(典型偶氮类染料,C18H13N3Na2O8S2,CAS号3734-67-6,简写为ARG)进行电催化氧化降解,考察电流密度、盐的质量分数和pH值对其电化学降解性能的影响。
1 实验方法
1.1 材料、试剂和仪器
钛网,质量分数为99.6%,购自宝鸡钛业有限公司;Ti4O7粉末,60~100目,购自河南龙兴钛业股份有限公司;ARG,分析纯,购自International Laboratory(USA);其余实验药品均为分析纯,购自国药集团,使用前未做进一步纯化处理;去离子水,由南京易普易达EPET-40TF型纯水机制得。
1.2 Ti4O7阳极制备
等离子喷涂法制备Ti4O7阳极流程如下。①Ti网基体预处理:采用粗砂纸和细砂纸先后对钛基体表面进行打磨处理;配制体积比为1∶1的NaOH和丙酮混合溶液,将钛网置于其中,加热至85 ℃处理30~60 min;清洗后将其置于微沸状态下质量分数为10%的草酸溶液中2 h,清洗后待用。②使用等离子喷涂设备喷涂熔融的Ti4O7颗粒制备功能涂层:采用等离子体炬(Saint-Gobain ProPlasma STD,内部温度范围为10 000~15 000 ℃)将60~100目Ti4O7粒子熔化后,以氩气为载气,喷速为30 g/min,喷油器直径为1.8 mm,注入角度为+10°,喷射到经过喷砂预处理的钛网上,从而形成粗糙的表面,得到均匀连续的层状涂层。
1.3 Ti4O7阳极性能表征
采用扫描电子显微镜(EVO 10型钨灯丝扫描电镜,德国卡尔蔡司公司)、X射线衍射(Bruker D8 Advance型,德国布鲁克公司)、X射线光电子能谱(AXIS Ultra Dld型,英国Kratos公司)对阳极的表面形貌进行表征。
采用电化学工作站(CHI660D型,上海辰华仪器公司,三电极体系。参比电极为Ag/AgCl(饱和KCl),对电极为铂片,工作电极为制备的电极)进行电化学测试。测试条件如下:0.5 mol/L Na2SO4溶液,扫描范围为0~2.0 V,扫描速度为10 mV/s;电化学交流阻抗测试的扫描范围为105~0.01 Hz,振幅为5 mV。
1.4 强化寿命实验
电极的强化寿命实验选取两电极体系进行测试,电源为KRS-3010直流电源,工作电极为制备的Ti4O7电极(1 cm×1 cm),阴极为相同尺寸的钛片。在强酸性条件(0.5 mol/L的H2SO4)下通电电解,电流密度为1 000 mA/cm2。观察并记录槽电压,当槽电压达到10 V时,即认为电极失效。
为了测试电极在强化寿命实验中表层氧化物是否有溶解情况,对强化寿命实验结束时的电解液液以0.45 μm膜进行过滤,随后采用ICPE-9000型电感耦合等离子体(ICP)光谱仪(日本岛津公司)进行ICP测试。
1.5 电催化降解实验
实验中所使用的电催化氧化体系均为“阴-阳-阴-阳”四电极体系。其中,阳极为制备的Ti4O7网状电极,阴极为相同尺寸的钛网,电极间距为10 mm,电解质为0.1 mol/L的Na2SO4(高盐体系下的溶液电解质为质量比1∶1的NaCl和Na2SO4),磁力搅拌器转速为200 r/min,目标有机物为酸性红G,溶液体积为500 mL,每隔一定时间取10 mL样品,进行相关指标的测试分析。每组实验均重复进行3次,取平均值作为实验结果。
1.6 分析方法
1.6.1 X射线衍射测定Ti4O7相对含量
在多相混合物中,任意两相的衍射强度比与其他物相的含量无关。X射线衍射测定中选取α-Al2O3为标准参比物质,求某物相与α-Al2O3等含量时二者最强峰的强度比,称为此物相的K值。若某两相的K值均可知,则此两相等含量时的最强峰强度比可知。多相混合物中,若每个相的K值可知、全部物相的含量为100%,则各相含量比可知。K值法最主要的优点是无需做出工作曲线,只需扫描待测样品即可[17]。
基于以上理论,对于亚氧化钛粉末/电极多相混合系统,系统中存在Ti4O7、TiO2、Ti5O9等N个相。选定Ti4O7相为内标物,通过 PDF 卡片查到每个相的K值,就可以计算出以其中的Ti4O7相为内标物时样品中每个相的K值。X相的质量分数可以表示为

(1)
式中:i=1表示选定的Ti4O7相,即A相;i=1,…,N,N表示样品中有N个相。
1.6.2 水样分析方法
采用紫外分光光度计(UV2600A型,上海尤尼科公司)选取波长λ=505 nm的紫外线测试溶液中的ARG含量,采用COD快速测定仪(ET 125 SC型,德国罗威邦公司)、总有机碳(TOC)分析仪(ET1020A型,欧陆科仪供公司)测定溶液COD、TOC值。
根据测试数据计算脱色率(ηcolor)、COD去除率(ηCOD)、TOC去除率(ηTOC),进而计算平均电流利用效率(ηACE,%)和降解COD所需的单位能耗(DCOD,kW·h/g),计算式为
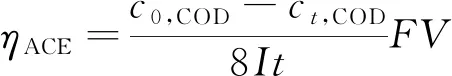
(2)
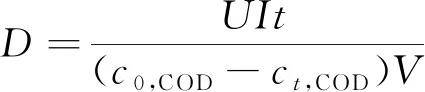
(3)
式中:c0,COD表示初始时刻水样COD的含量,mg/L;ct,COD表示t时刻水样COD的含量,mg/L;F表示法拉第常数;V表示溶液体积,m3;I表示电流,A;t表示处理时间,s;U表示电压,V。
2 结果与讨论
2.1 Ti4O7阳极的材料性能表征
2.1.1 形貌分析
图1为Ti4O7粉末和阳极在不同放大倍数下的SEM图。可以看出,实验所采用的Ti4O7粉末呈空心球状多孔结构(图1(a)),制备后电极呈均匀连续涂层(图1(b)、(c))。在较低放大倍数(100倍)下,可观察到电极表面粗糙,有不规则形状的颗粒,整体呈蜂窝状分布,结构致密,没有明显裂痕(图1(b));在较高放大倍数下(2 500倍),可清楚地观察到阳极表面分布着丰富的颗粒孔隙结构,颗粒均为堆叠状,层层累积而成(图1(c))。传统的热分解法制备电极时,由于涂层和钛基体的热膨胀系数不一致,导致表面涂层不可避免的存在大量裂纹[18],使得电解液可以经此途径向钛基体方向渗透,由此严重影响电极的稳定性和使用寿命[19]。与文献[20]相比,本文所制得的电极表面裂纹数量非常少,且Ti4O7与钛基底的热膨胀系数接近,这将十分有利于该电极在实际使用过程中的稳定性能。该结果也与Teng等[21]研究成果一致。

(a)Ti4O7粉末 ×2 000

(b)Ti4O7阳极 ×100

(c)Ti4O7阳极 ×2 500
2.1.2 XRD结果分析
尽管钛氧化物所有Magneli相晶体结构差异不大,经常会发生衍射峰的相互重叠[22],但利用XRD对其进行检测,并将所获图谱与标准谱图进行比对分析,即可明确具体的晶体结构。图2为制备原料Ti4O7粉末与电极的XRD对比图谱。可以看出,在2θ分别为20.74°、26.35°、31.82°、35.60°、40.58°、55.05°、63.77°和66.43°等衍射角处,都出现了特征衍射峰,且峰高明显,符合XRD标准谱图(JCPDF no.77-1392)中Ti4O7的特征衍射峰,晶面分别为(120)、(11-5)、(040)、(224),可以确定实验采用的粉末与制备电极表面的物质均为Ti4O7相。
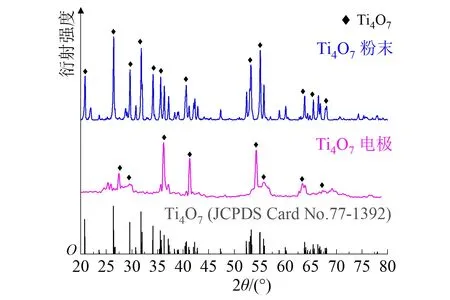
图2 Ti4O7粉末和阳极的XRD谱图Fig.2 XRD patterns of Ti4O7 powder and anode
对得到的粉末和电极XRD图谱进行物相鉴定,根据不同物相的K值和最强峰强度,按照1.6.1小节计算相含量的原理,利用式(1)得到亚氧化钛粉末Ti4O7相含量为92.56%,电极表面Ti4O7相含量为56.34%。计算结果表明:实验所采用的商用Ti4O7粉末物相纯洁;与粉末相比,电极制成后其表面涂层中的Ti4O7相所占比例减小。这可能是因为实际制备过程中瞬间高温条件下部分亚氧化钛粉末物相发生改变或直接被氧化为不导电的二氧化钛,故应注意在喷涂过程中应高度重视还原性气氛保护,进一步提高制成电极中Ti4O7相所占比例,确保电极性能的优异稳定。
2.1.3 X射线光电子能谱分析
图3为Ti4O7阳极的XPS图。由图3(a)可知,电极的构成元素为钛元素和氧元素。对Ti 2p轨道进行分析(如图3(b)所示)可知,钛元素峰值所对应的结合能分别为Ti 2p3/2(458.5 eV)和Ti 2p1/2(463.9 eV),对光谱进行分峰和高斯拟合可知,电极表面钛元素的化学价态主要以Ti4+和Ti3+的混合形式存在[23]。同上,对O 1s轨道进行分析(如图3(c)所示)所示)可知,氧元素主要以O2-(529.9 eV)、OH-(531.9 eV)、H2O(533.2 eV)3种形式存在,其中O2-为强结合态的晶格氧,OH-为吸附羟基氧,H2O为羟基吸附的水[24]。吸附羟基氧在电极表面可以转化为氧化能力极强、活性极高的羟基自由基,对有机污染物有更高的催化活性[25-26]。
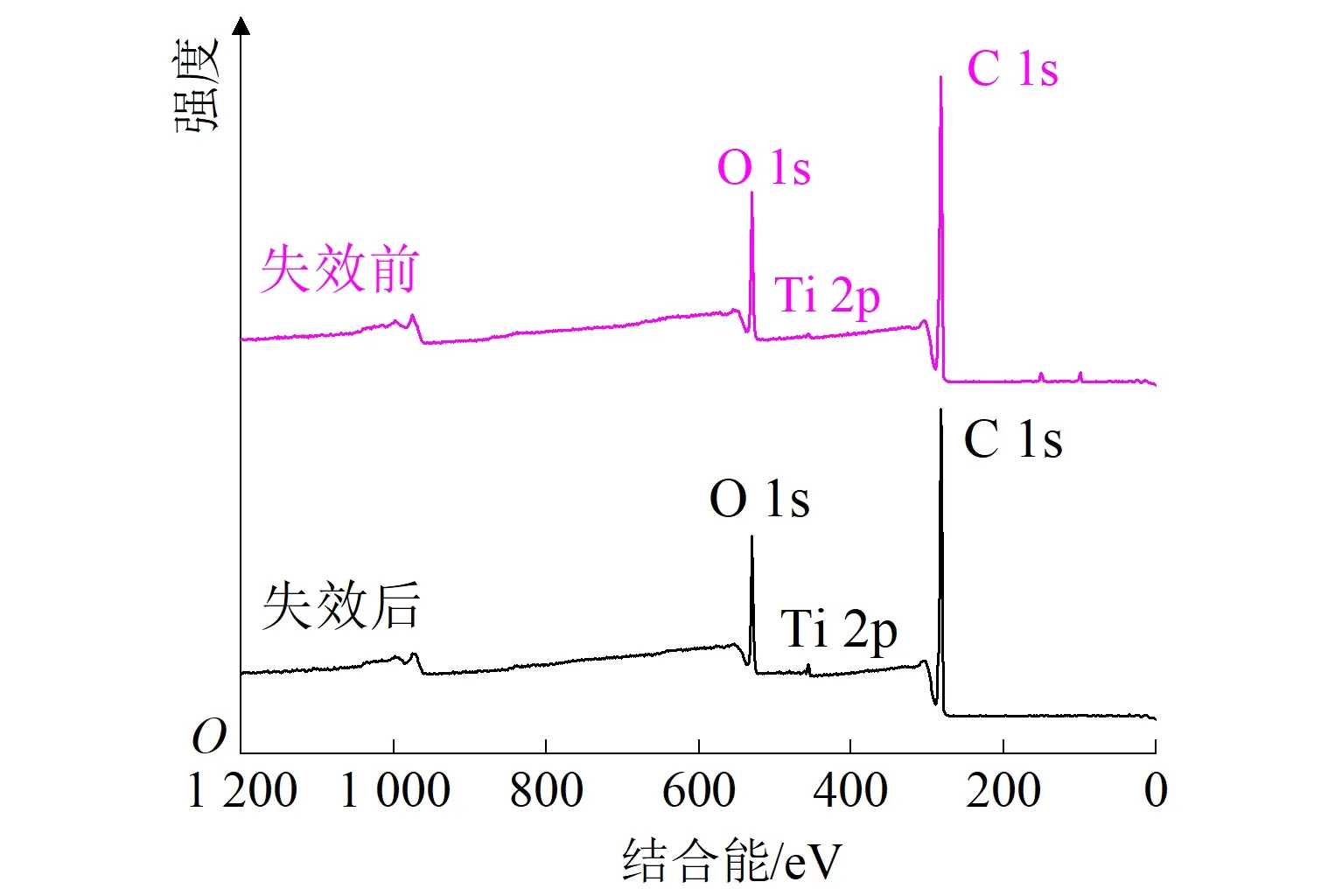
(a)XPS全谱扫描
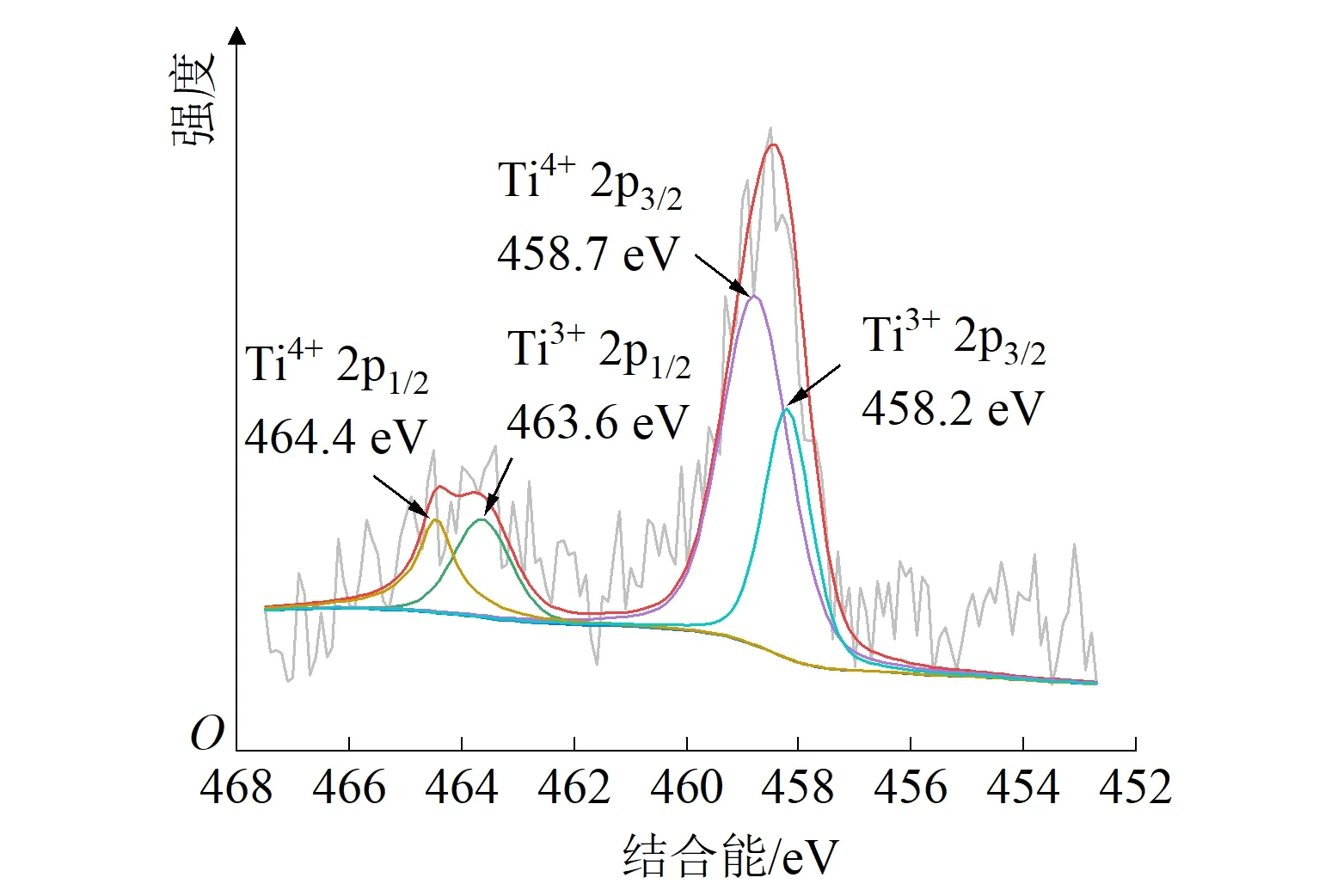
(b)Ti 2p轨道光谱

(c)O 1s轨道光谱
2.2 Ti4O7阳极的电化学性能评价
对Ti4O7阳极进行了150圈的循环伏安扫描,结果如图4(a)所示。可以看出,该种电极析氧过电位约为1.497 V(vs Ag/AgCl参比电极)。第1圈、第100圈和第150圈的3条曲线的吻合度较高,说明该电极经历多次循环后性质基本不变,具备较高的电化学稳定性。Ti4O7阳极优越的电化学性能得益于其独特的晶格结构。Ti4O7是由八面体结构的TiO2二维尺寸链组成的网状结构[27],每3层TiO2之间通过表面互相接触与反应共用一个TiO层,两侧的TiO2层可以包裹其导电带,使其发生晶体结构崩塌和改变的可能性较小,也决定了该电极拥有良好的抗腐蚀性和稳定性[22]。
图4(b)为Ti4O7阳极的电化学阻抗谱图。通过实验拟合数据可知,Ti4O7阳极的欧姆内阻,即曲线与X轴的交点为3.876 Ω,说明其具备高的导电性;电荷转移内阻为2 462 Ω,可能在实际制备过程中,部分亚氧化钛粉末被氧化为不导电的二氧化钛,
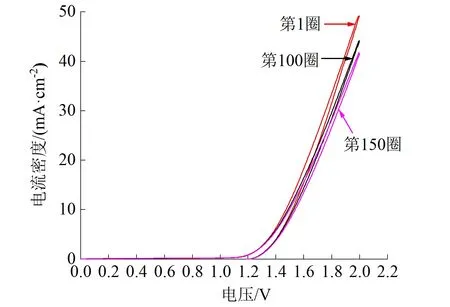
(a)循环伏安曲线
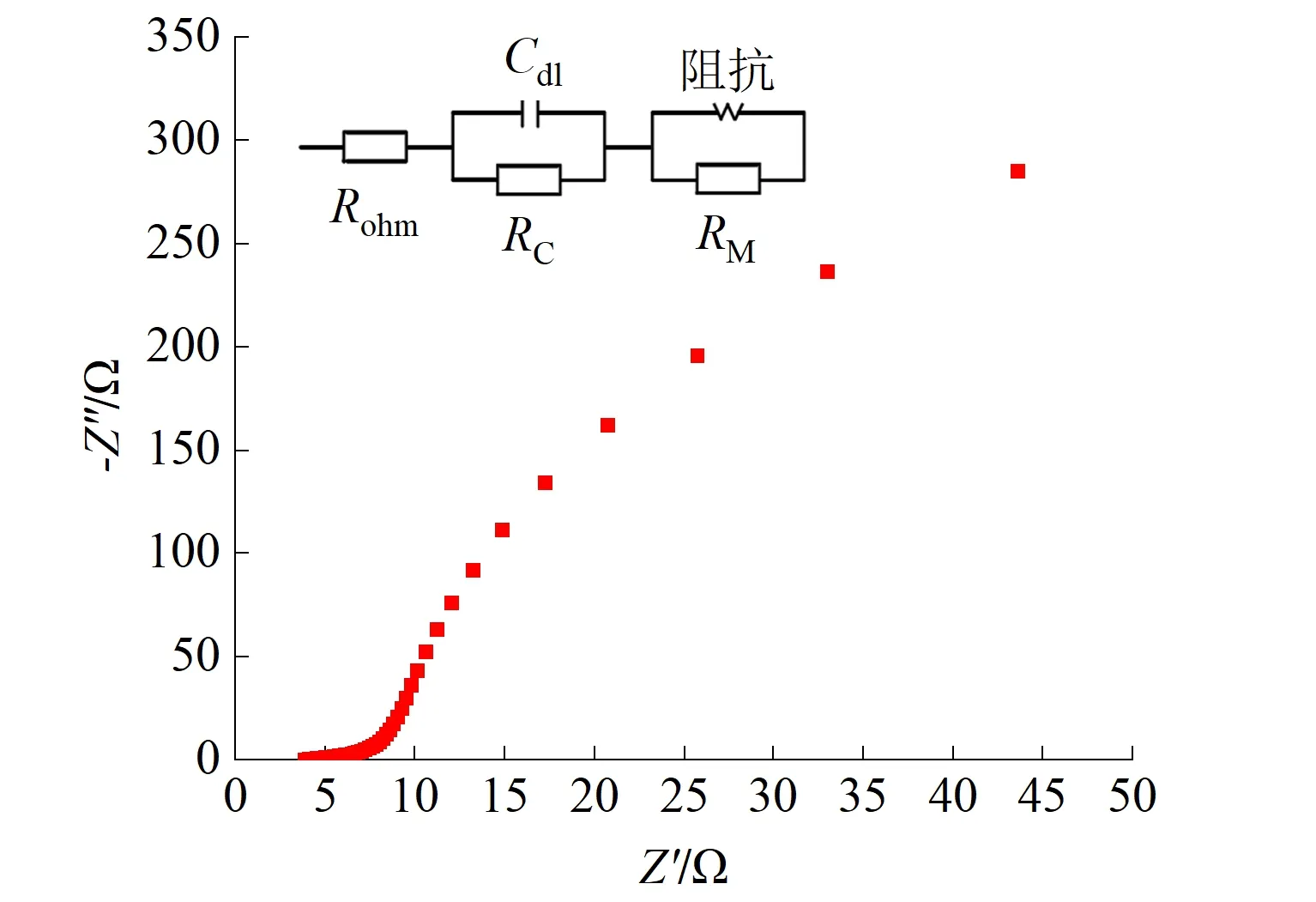
(b)电化学阻抗谱(小图为等效电路图)
Ti4O7anode
造成电极表面电荷转移内阻较高,这与XRD计算结果一致;扩散内阻为6.935 Ω,说明在电化学反应过程中,溶液中的溶质能够快速在电极表面扩散和反应,提高了电化学反应速度。因此,Ti4O7阳极具备较小的电阻和较高的电催化氧化活性,能够有效地降解有机污染物。
2.3 高盐体系下Ti4O7阳极电催化氧化性能
2.3.1 不同因素对高盐体系下ARG电催化降解的影响
为了评价Ti4O7阳极在高盐体系下的电催化活性,采用不同电流密度对ARG模拟废水进行了催化降解实验,结果如表1和图5所示。由表1可知,在120 min时,随着电流密度的增大,脱色率由99.93%提高至99.97%,近乎完全褪色,且在20 min时,脱色率就高达99.57%以上,表明ARG染料在短时间内就可以得到有效分解。从图5(a)可以看出,120 min时,10、15、20 mA/cm2电流密度下COD去除率分别为46.86%、71.88%和100.00%。对3个电流密度下的COD去除率数据进行了动力学拟合,发现它们基本符合准一阶动力学过程,如图5(b)所示,COD去除反应的速率常数分别为0.005 4 min-1(R2=0872 1)、0.011 0 min-1(R2=0.988 1)和0.023 3 min-1(R2=0.963 0)。此外,通过表1可知,随着电流密度增加:TOC去除率由16.25%提高至19.34%,平均电流效率由9.86%增加至10.52%,基本维持稳定;DCOD由0.11 kW·h/g增加至0.14 kW·h/g。
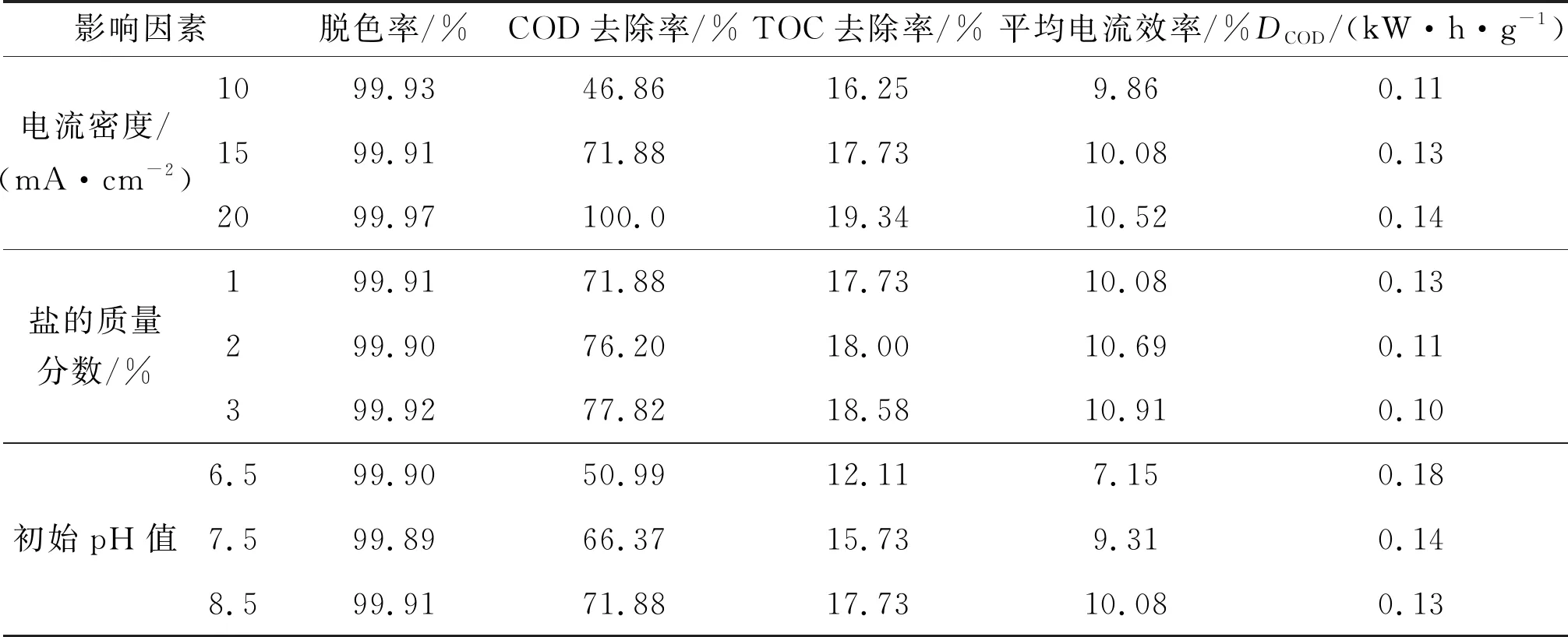
表1 不同条件下120 min内Ti4O7阳极对高盐ARG的电化学氧化降解效果
高盐体系下的电催化氧化过程中,随着电流密度的增大,参与反应的电子越多,生成的·OH越多[28],更多的Cl-可被氧化为HClO和ClO-等强氧化态物质[29],二者协助氧化ARG,造成ARG的迅速氧化脱色、COD去除率的大幅度提升以及TOC去除率的小幅度提升,且此时溶液中有足够的Cl-与电子发生反应,导致电流利用效率基本维持稳定。但是,电化学过程中伴随的欧姆加热过程使平均能耗增加。因此,采用适合的电流密度(15 mA/cm2)时,既能保证高效降解ARG,又能将能耗降到最低。
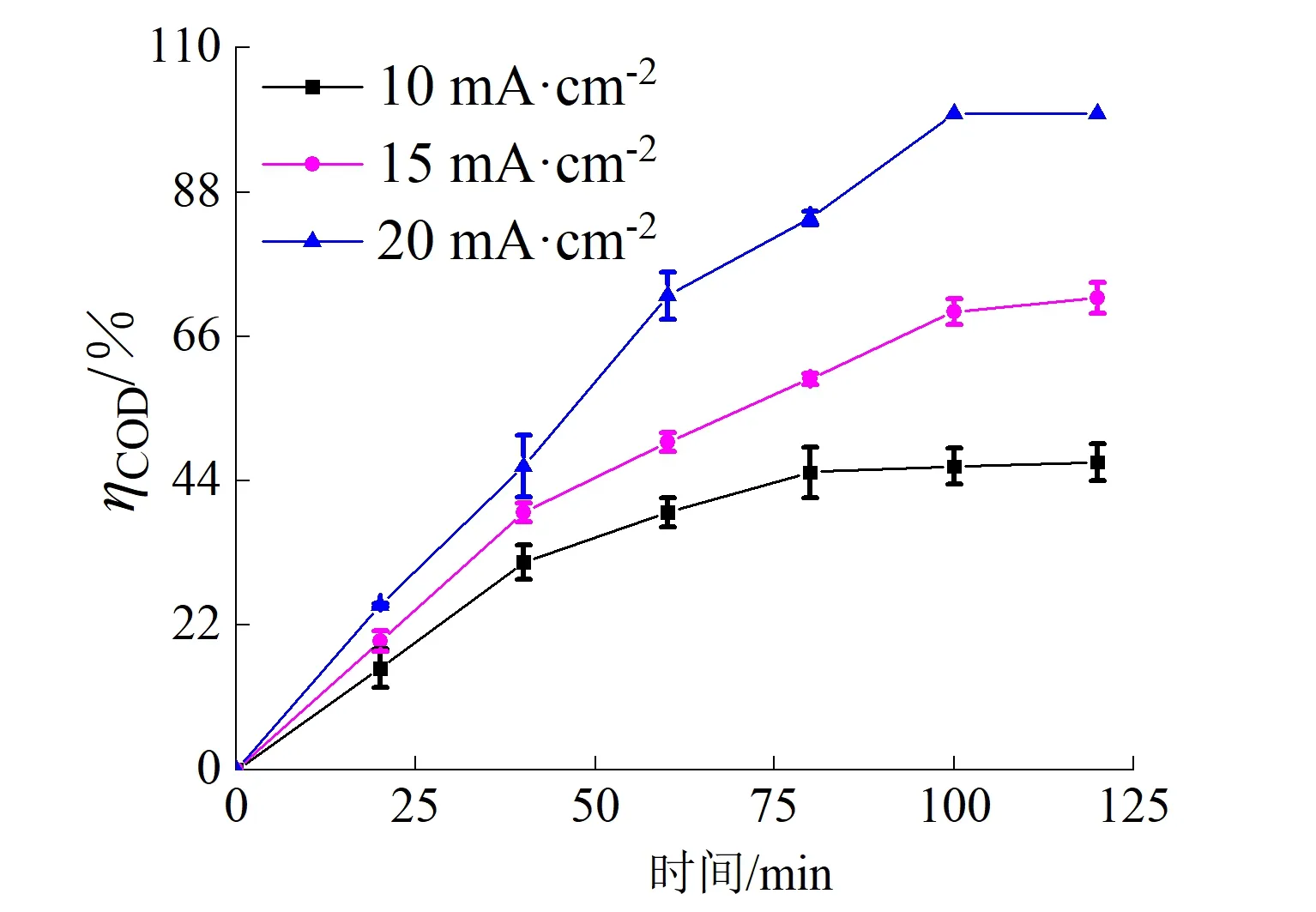
(a)COD去除率
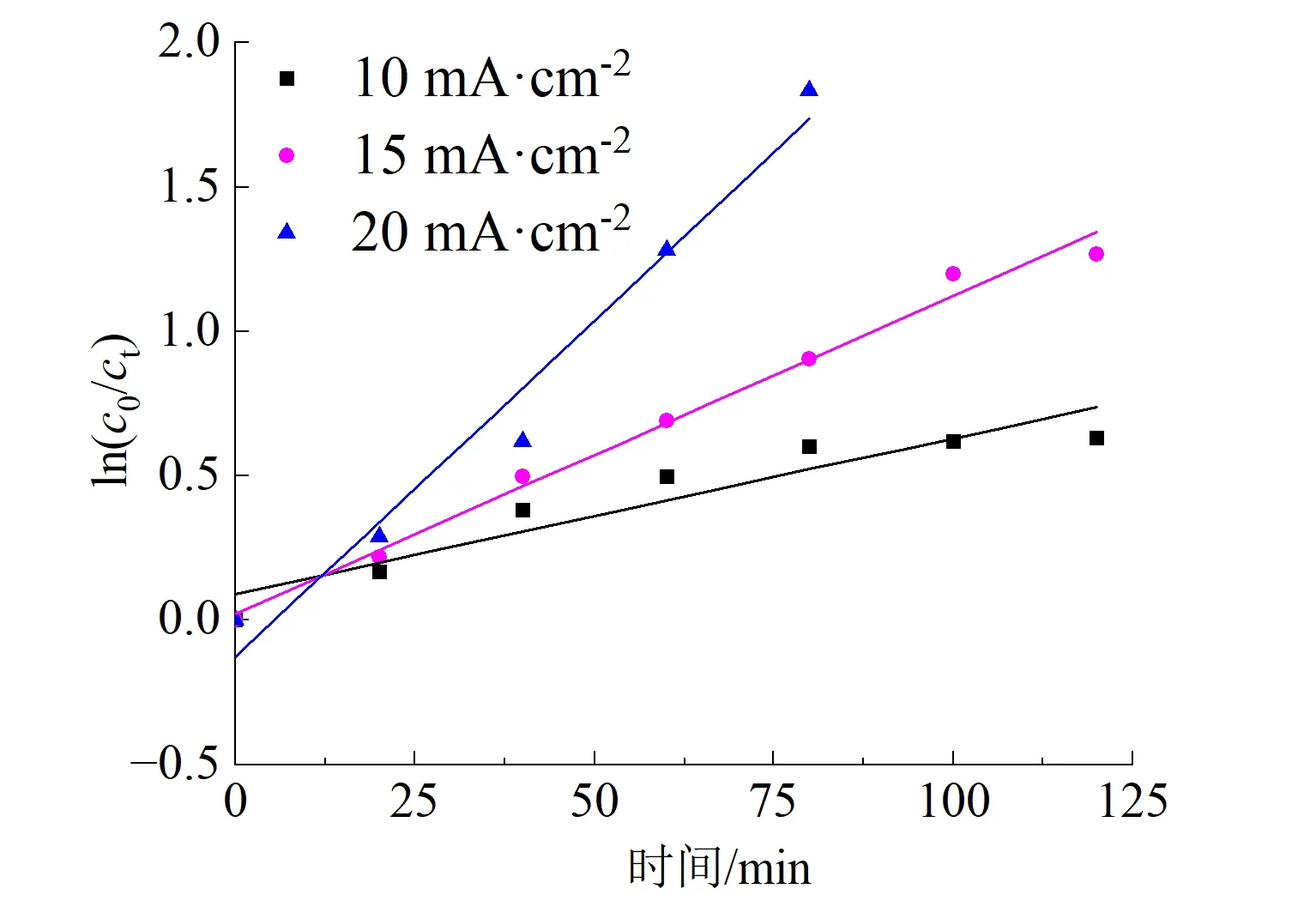
(b)动力学拟合
此外,本文还考查了不同盐的质量分数(1%、2%和3%)以及不同初始pH值(6.5、7.5和8.5)对ARG脱色率、COD去除率、TOC去除率、平均电流效率及能耗的影响,结果如表1所示。由表1可知,在高盐体系下,增加盐的质量分数对ARG的降解效果没有显著提升,平均电流效率以及能耗的变化幅度都不明显。这是因为当盐的质量分数过高时,易在电极表面形成盐膜,阻碍污染物向电极表面传质,降低·OH对污染物的有效去除[30],导致去除效率增幅较小。与此同时,过高含量的盐在电化学过程中会产生大量剧毒的Cl2,对周围环境及实验人员造成一定的危害[19]。随着初始pH值的增大,COD去除率、TOC去除率、平均电流效率均有所提高,能耗有所下降。此现象可以解释为较多的氢氧根离子可以促进羟基自由基的产生,对ARG产生催化降解作用[31]。此外,尽管ARG在其他pH值条件下降解效果有所下降,但是依然具有一定的降解能力,证实Ti4O7阳极的催化氧化能力能在一个较宽的pH值范围内起作用。对比以Ti/Ta2O5-IrO2、石墨及Ti/RuO2-IrO2电极作为阳极处理高盐废水的效果(COD去除率分别为62.0%、43.7%及49.6%)[16]可知,Ti4O7阳极有着明显的优越性。
2.3.2 高盐体系下ARG电催化降解原理推测
根据之前工作可知,ARG在电催化氧化体系中是与电极表面产生的羟基自由基发生反应而被降解矿化的[32],而在高盐体系下由于Cl-在电化学过程中会被氧化为HClO和ClO-等强氧化态物质(见式(6)、(7)),这些强氧化性物质会协助羟基自由基氧化ARG,避免了阳极表面聚合物膜的形成,提高了降解效果[33],故推测在高盐体系下其阴、阳极反应如下,机理如图6所示。

图6 高盐体系下ARG电催化降解原理推测Fig.6 Theory of ARG electrocatalytic degradation in high salt system
阳极反应为
2H2O→O2+4H++4e-
(4)
2Cl-→Cl2↑+2e-
(5)
Cl2+H2O→HCl+HClO
(6)
HClO→H++ClO-
(7)
阴极反应为
2H2O+2e-→H2+2OH-
(8)
Mn++ne-→M
(9)
2.4 Ti4O7阳极的稳定性分析
2.4.1 强化寿命测试
图7为Ti4O7阳极强化寿命测试结果,实验共计约350 h,实验过程中溶液的颜色由无色透明逐渐变为红褐色,且存在絮状物。如图7所示,电极整体失效过程可分为3个阶段。①活化期:在0~48 h内,电极处于活化阶段,由于电解质未能与电极表面充分接触,同时电极表面存在一些杂质,导致电阻较大,初始槽压较高,随着电解的进行,杂质逐渐脱落,表面电阻减小,槽压逐渐下降。②稳定期:在48~320 h内,电极处于稳定阶段,说明此时电解质与电极表面充分接触,电极表面涂层稳定,没有大块脱落,电极电阻基本不变,槽压稳定。③失效期:在320 h以后,槽压开始上升,在350 h时陡升至10 V,认定此刻电极已经损坏失效,整体的失效过程与黄国胜等[34]的研究结果一致。

图7 槽压随电解时间的变化Fig.7 Variation of tank pressure with electrolytic time
2.4.2 失效电极性质表征
为进一步分析电极失效原因,对电解液进行ICP测试。结果显示,溶液中的Ti元素含量为1 458 mg·L-1,由此推测在强化寿命过程中,电极表面的亚氧化钛涂层是在逐渐溶解的。
对失效电极进行SEM、XPS和XRD表征,结果如图8所示。图8(a)为失效后电极表面的SEM图(放大100倍),与失效前相比,电极表面涂层凸起并产生明显裂痕,裂痕会造成电极内部产生内应力,表层和底层出现明显的界面应力,导致活性层出现鼓泡、脱落,电极的快速失活以及电催化能力的下降[35]。由图3(a)可知,失效前后电极表面的主要元素构成不变,均为钛元素和氧元素。图8(b)、(c)表明:失效后电极表面钛元素的峰位于Ti 2p3/2(458.5 eV)和Ti 2p1/2(464.2 eV)处,由Ti3+和Ti4+的混合形式变为单一Ti4+[23];氧元素仍以O2-(529.9 eV)、OH-(531.9 eV)、H2O(533.2 eV)的形式存在,O2-的相对含量提高了,可推测在电极氧化过程中,产生的新生态氧进入到了晶格内部,将表面的亚氧化钛氧化为不导电的TiO2,或者与钛基体反应生成不导电的TiO2,最终导致电极失效。
为进一步确定失效后电极表面的晶型,对其进行XRD测试,结果如图8(d)所示。可以看出,在2θ分别为27.4°、36.0°、41.2°、54.3°、和64.0°等衍射角出处都出现了特征衍射峰,且峰高明显,符合XRD标准谱图(PDF-#99-009-FOM39.9)中TiO2的特征衍射峰,晶面分别为(110)、(101)、(111)、(211)、(310),故电极表面的物质为TiO2,推测失效原因为表面Ti4O7被氧化为TiO2。

(a)失效后的SEM图
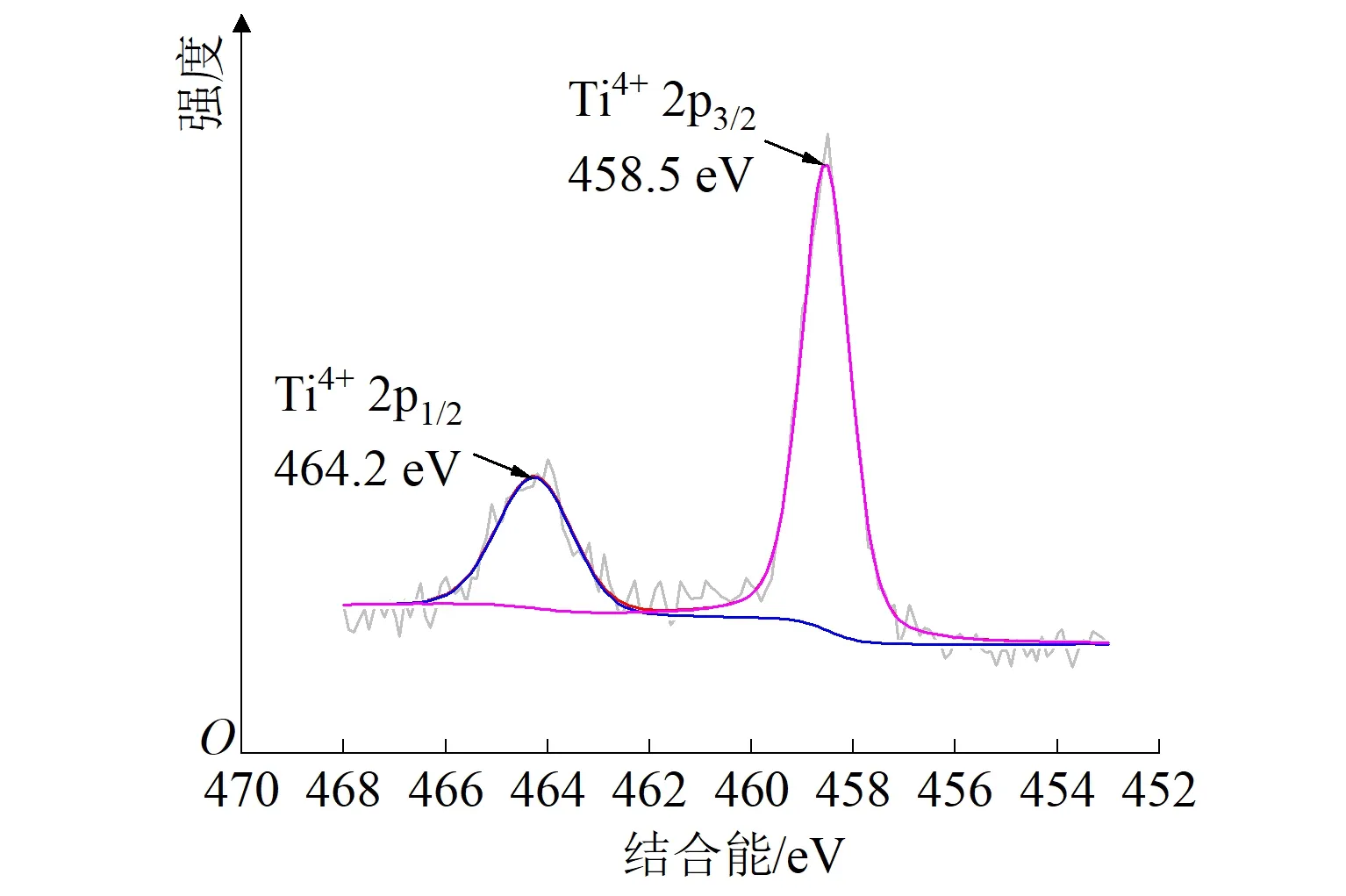
(b)失效后的Ti 2p轨道光谱

(c)失效后的O 1s轨道光谱
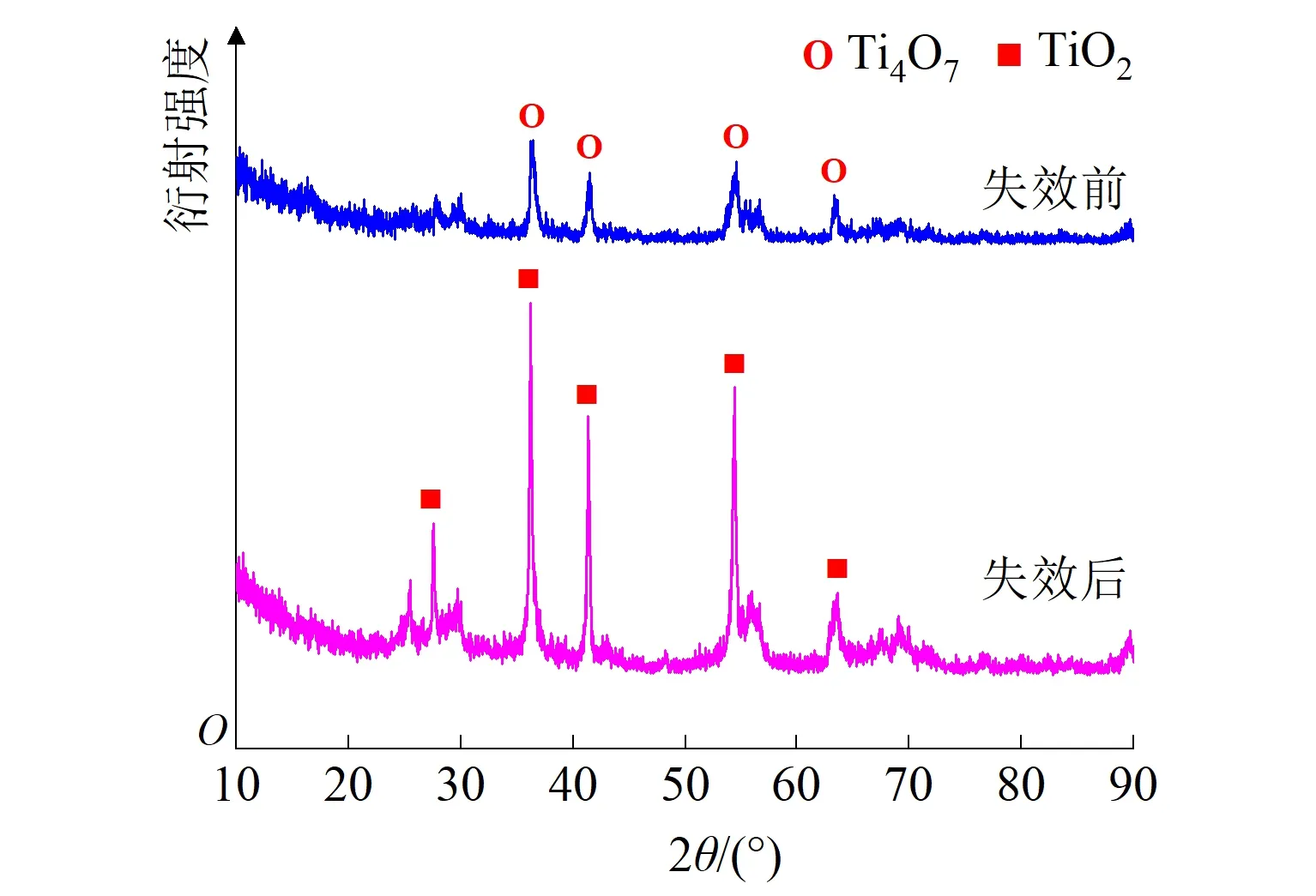
(d)失效前后的XRD对比
2.4.3 亚氧化钛电极失效机理推测
传统钛基金属氧化物电极的失效机理,国内外有很多文献报道,目前得到广泛认可的是活性成分的溶解和钛基体钝化两种[36]。与之相类似,亚氧化钛电极失效也存在涂层脱落与基体钝化的情况,不同的是金属氧化物电极的涂层溶解更多的是以离子的形式扩散到溶液中,而Ti4O7阳极因其稳定的化学特性,常以机械破损、氧化失活的形式失去活性。
结合以上分析,推测亚氧化钛电极失效的过程及原因具体为以下3条:①电极氧化过程中,表面涂层会不断溶解直至产生一定程度的裂纹,电解液会渗入裂纹处并不断发生电化学反应,产生大量新生态氧,释放出氧气,在狭小的空间内迅速释放的氧气会不断冲击基体和表面涂层,造成表面涂层裂纹不断扩大直至发生剥离或脱落;②电极氧化过程产生的新生态氧通过表面裂痕朝钛基体方向扩散,在该过程中不断与钛材料反应,使其由导电性能良好的Ti4O7相转变其它导电性能差的物相,直至最终转变为不导电的TiO2,导致表面活性层失效;③新生态氧扩散到钛基体之后,会使其被氧化为不导电的TiO2,导致电极基体和活性层中间形成绝缘隔离带,致使电极失效。
3 结 论
本文以新型亚氧化钛电极作为阳极,进行了其在电催化氧化降解高盐有机污染物废水中的影响因素研究,并进行了强化寿命测试,得出以下主要结论。
(1)实验采用等离子喷涂法成功制备了Ti4O7阳极,并对其进行SEM、EDS、XPS、XRD及电化学性能等表征,发现此电极表面具备丰富的孔洞结构,具有优异的传质能力。同时,Ti4O7阳极具备较高的析氧过电位、优异的导电性能和良好稳定性等优点。
(2)实验采用Ti4O7阳极对高盐体系下ARG进行电催化降解,在实验条件电流密度为15 mA/cm2、盐的质量分数为1%、初始pH值为8.5时得到最佳降解效果,120 min内ARG脱色率为99.91%,COD去除率为71.88%,降解效果受含盐量、pH值等因素影响较小,证实了Ti4O7阳极催化能力的广泛适用性。
(3)通过对电极进行强化寿命测试可知,其强化寿命时间约为350 h,具备优异的稳定性能。分析其失效过程及机制为:表面涂层在电解过程中部分溶解进入电解液,部分被缓慢氧化为绝缘态的TiO2。此外,表面涂层在析氧反应产生气体的剧烈冲击下出现破损脱落,导致部分钛基体被钝化为不导电的TiO2。
猜你喜欢
化工管理(2022年14期)2022-12-02 11:44:06
保健文汇(2022年4期)2022-06-01 10:06:50
陶瓷学报(2021年1期)2021-04-13 01:33:38
超硬材料工程(2019年3期)2019-09-06 02:12:20
中国有色金属学报(2018年2期)2018-03-26 07:58:37
中南大学学报(自然科学版)(2016年2期)2017-01-19 07:37:25
材料科学与工程学报(2016年1期)2017-01-15 13:33:40
中国资源综合利用(2016年7期)2016-02-03 03:00:13
船舶标准化工程师(2015年5期)2015-12-03 11:00:28
无机化学学报(2014年7期)2014-02-28 17:32:26
