
常染色体隐性遗传性Alport综合征误诊为IgA肾病1例报道并文献复习
2023-05-04 09:18袁平郑莎莎
中国生育健康杂志 2023年3期
袁平 郑莎莎
病例资料
患儿,女,7岁,主因“间断血尿、蛋白尿2年余”于2021年01月25日入本院。2018年11月06日患儿因“上呼吸道感染”4 d后出现尿色异常就医,表现为全程深红色尿,无尿频、尿急、尿痛,无尿中沉渣及絮状物,无水肿、少尿。当地医院检查:尿常规示红细胞3101个/uL,尿蛋白2+;24 h尿蛋白定量0.337 g/d(19 mg/kg);补体C3、C4正常;ASO增高(583.96 IU/mL);肾功能(血肌酐31 μmol/L)、自身抗体谱、ANCA无异常。病程第5天当地完善肾穿活检,病理结果免疫荧光:IgA 3+、IgG 2+、IgM 1+、C3 2+、C1q 1+、Fib -,IV型胶原α3、α5链染色肾小球基底膜和包曼氏囊均为阳性。光镜:可见9个肾小球,未见肾小球球性硬化及节段性硬化,六胺银(PASM)染色可见5个肾小球,未见肾小球球性硬化及节段性硬化;肾小球系膜细胞和基质轻度弥漫性增生,系膜区少许嗜复红蛋白沉积,毛细血管襻开放,基底膜无明显增厚,无钉突双轨改变,无系膜插入,上皮下、内皮下无明显嗜复红蛋白沉积,未见白金耳样结构,壁层上皮细胞无增生,未见新月体形成;肾小管上皮细胞颗粒变性,无明显萎缩,肾间质无明显炎症细胞浸润及纤维化,小动脉管壁无明显病变。电镜:肾小球基底膜弥漫性偏薄,厚度小于250 nm,足突部分融合,系膜区可见电子致密物沉积。病理诊断:轻度系膜增生性IgA肾病(M1E0S0T0)。结合肾脏病理,临床诊断为IgA肾病(IgA nephropathy,IgAN),给予甲泼尼龙冲击1疗程,继以醋酸泼尼松35 mg/d(2 mg/kg/d)口服并递减,联合赛可平250 mg Bid(30 mg/kg/d)、洛汀新5 mg Qd治疗2年余,期间复查尿常规蛋白1+~2+,尿红细胞30~满视野/HP;尿蛋白定量0.24~0.31g/24 h。肾功能正常。现为进一步诊治,特来本院。目前口服醋酸泼尼松10 mg/d,赛可平和洛汀新剂量同前。
既往、个人史无特殊。家族中有一哥哥,现年10岁,体健,未查过尿常规;父母体健,未查过尿常规。
入院查体:神清,精神反应好,轻度库欣貌,咽无充血,扁桃体无肿大,心肺腹无异常,神经系统未见阳性体征。
入院后主要辅助检查:全血生化示肌酐35.90 μmol/L,肝功能、电解质、心肌酶谱均正常;尿常规示蛋白质1+,红细胞40~50/HP;尿蛋白/尿肌酐0.38 g/gcr;肾早期损伤指标示尿微量白蛋白60.90 mg/L,尿转铁蛋白和球蛋白正常;24 h尿蛋白定量 0.23 g;校正24 h肌酐清除率140 mL/min/1.73 m2;iPTH 14.45 pg/mL,25-OH-VitD 27.61 nmol/L;泌尿系超声示双肾实质回声稍增强,肝胆胰脾未见异常,未探及腹腔积液。眼科会诊示患儿双眼视网膜颞侧变薄,符合Alport综合征(Alport syndrome,AS)眼部特征。耳鼻喉科行纯音测听提示双耳大致正常听力。患儿哥哥及父母尿常规蛋白均阴性,红细胞3~5/HP。结合患儿临床过程,按IgA肾病治疗效果不佳,肾组织电镜示肾小球基底膜弥漫性偏薄,家族中哥哥及父母均有镜下血尿,考虑遗传性肾小球肾炎不除外,二代测序完善家系遗传性肾脏病相关基因变异分析,结果回报COL4A4基因存在2个位点杂合变异,c.5044C>G(p.R1682G,父源)和c.871-1G>A(母源),按照美国医学遗传学与基因组学学会(The American College of Medical Genetics and Genomics,ACMG)指南,变异位点分别初步判定为致病意义未明(PM2 +PM5+PP3)及疑似致病性变异(PVS+PM2),结合患儿临床表现、家族史及药物反应,明确诊断为“常染色体隐性遗传性Alport综合征”,见图1、图2。

图1 患儿家系的COL4A4基因测序图1注:a为变异位点1,chr2:227872070,NM_0000 92;exon48,c.5044C>G(p.R1682G),为错义变异
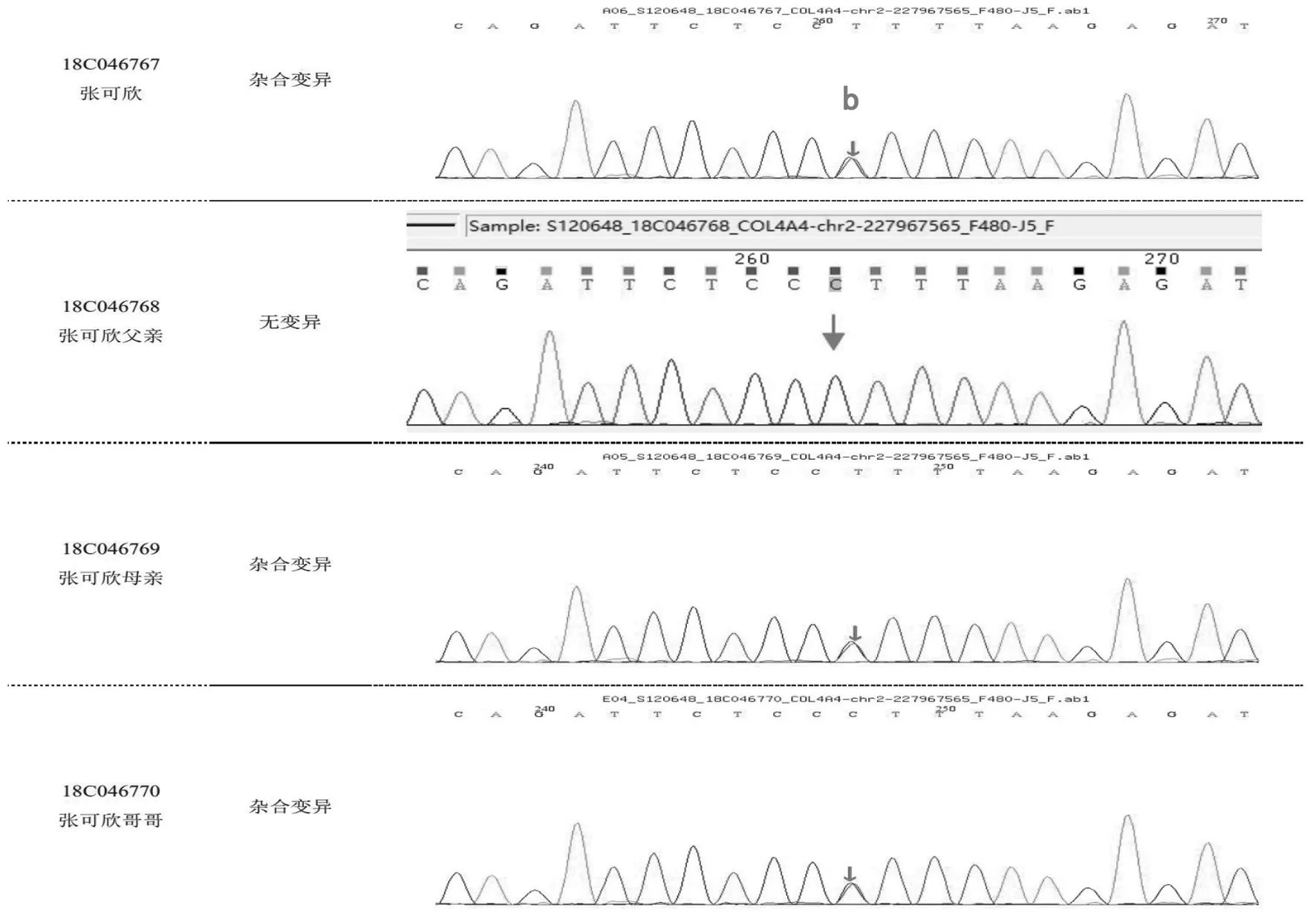
图2 患儿家系的COL4A4基因测序图变异位点2注:b为变异位点2,chr2:227872070,NM_0000 92;exon48,c.5044C>G(p.R1682G),为错义变异。峰图显示的碱基有可能为被检测碱基的反向互补序列,如:c.163G>A,峰图可显示为 G>A 或其反向互补序列 C>T
患儿诊断明确后,逐渐减停激素、赛可平,继续洛汀新5 mg Qd口服,随访近1年,24 h尿蛋白定量(0.25~0.38 g)和肾功能(血肌酐32~36 μmol/L)较前无明显变化,生长发育同正常同龄儿。
讨论
IgAN是儿童最常见的原发性肾小球疾病之一,也是成人慢性肾脏疾病及终末期肾脏病的重要病因之一,主要在青年中好发,儿童也很常见。儿童年发病率为(0.03~4.5)/100 000[1]。IgAN的主要病理学特征是免疫荧光以IgA在肾小球沉积为主以及局部炎症、系膜增生、肾小球纤维化等,其诊断基于主要依赖于肾组织免疫荧光染色结果。
AS是一种以血尿、蛋白尿和进行性肾功能衰竭为特征的慢性进展性肾脏疾病,可伴有高频感音神经性耳聋和眼部病变,其诊断主要依赖基因变异分析或肾组织电镜下肾小球基底膜特征性改变。IgAN和AS二者具有相同的临床表现(血尿、蛋白尿、终末期肾病),特别是二者也可合并发生,临床可能会发生误诊、漏诊,甚至过度用药误治的情况。
IgAN临床表现差异性大,起病形式包括无症状或轻微尿检异常(包括镜下血尿及少量蛋白尿)、持续蛋白尿及肾功能迅速恶化,一些患者没有进展到终末期肾脏病(ESRD),甚至有报道称一些患者自发缓解[2]。IgAN的经典表现是与黏膜感染相关的发作性肉眼血尿或持续性镜下血尿。儿童IgAN有和成人类似的蛋白尿水平,但肉眼血尿更常见,血压升高不明显,肾功能保留较好。儿童比成人更常出现肉眼血尿,通常与上呼吸道感染有关。AS是因编码Ⅳ型胶原蛋白α3、α4、α5链基因变异所致的遗传性肾脏疾病,在临床上表现为血尿、蛋白尿及进行性肾衰竭,部分患者合并耳聋和眼部改变[3],其遗传方式有X连锁显性、常染色体隐性、常染色体显性遗传,其中以X连锁显性最常见,约占85%,主要为COL4A5的基因缺陷导致,常染色体隐性遗传(ARAS)为COL4A3或 COL4A4基因缺陷所致,约占10%;常染色体显性遗传(ADAS),不到5 %,临床非常罕见[4]。该病起病隐匿,早期病变表现轻微,仅为镜下血尿,伴或不伴蛋白尿,肾功能异常多在学龄期甚至青春期后出现,听力和视力损害也同样出现较晚。
本例患儿以上呼吸道感染后发作性肉眼血尿起病,临床符合IgAN特点,结合病初肾脏病理免疫荧光以IgA沉积为主,符合IgAN诊断,但规律激素联合免疫抑制剂无明显疗效,尿蛋白定量无明显变化,不符合儿童IgAN诊断常规病程。进一步检查发现眼底AS特征性病变,且家族中哥哥、父母均镜下血尿,考虑遗传性肾小球肾炎不除外。基因变异证实COL4A4复合杂合变异(exon48:c.5044C>G p.R1682G,父源;exon15:c.871-1G>A,母源),诊断为常染色体隐性遗传AS(COL4A4复合杂合变异)。
IgAN合并AS国内有病例报道,X连锁显性遗传性AS多见[5-6]。已知正常健康人,肾组织活检免疫荧光IgA沉积者约10%~30%[7-8],因此其他一些其他常见遗传性肾小球肾炎通过肾组织活检可能被误诊为IgAN。本例患儿,临床经过激素联合免疫抑制剂治疗,尿蛋白水平无明显下降趋势,因此考虑其尿蛋白主要为原发病即常染色体隐性遗传AS引起,与肾组织IgA沉积无明显相关性,因此可以认为临床被误诊、误治或过度治疗。提示在临床工作中,以血尿起病的患儿很常见,既使通过肾活检明确诊断为IgAN,但如果在后期的治疗过程中出现病情多次反复或药物治疗效果欠佳,应积极询问家族史、完善家系成员尿常规筛查,必要时相关基因变异分析以明确诊断,有无合并遗传性肾小球肾炎或误诊可能。
AS的病理学常表现为系膜增生,时有IgA沉积,此外还有独特的肾小球基底膜(GBM)改变,包括基底膜变薄和/或板层化。然而,在IgAN中也经常观察到类似的GBM异常。这两种疾病在与病毒感染相关时也会出现血尿、蛋白尿,有时还会出现大量血尿。因此,即使根据临床和病理结果,也很难做出鉴别诊断。一项日本的回顾性研究[9]对5例同时伴有IgA沉积和GBM改变的患者进行了全面的基因筛查发现,其中4例被诊断为AS,1例为IgAN,伴有大量GBM改变,基因检测结果为阴性。在4例被诊断为AS的患者中,肾活检IgA在系膜区沉积在+至2+之间,未检测到肾小球Gd-IgA1沉积,尽管系膜区IgA呈阳性;在1例IgA肾病,IgA沉积呈3+,并观察到肾小球Gd-IgA1显著沉积。提示反复血尿,治疗效果不佳的病人,即使有典型的IgA肾病的临床特点及常规病理结果,也难区分IgAN还是AS,还需进一步行基因检测,以明确诊断AS。此外,Gd-IgA1染色有助于肾小球肾炎伴GBM异常和系膜IgA沉积的准确诊断,需要更多的分析来评估肾小球Gd-IgA1染色的效用。但国内绝大部分肾活检免疫荧光均未行Gd-IgA1染色,值得注意的是当反复血尿和/或蛋白尿的病人行肾活检免疫荧光提示IgA少量沉积时,需考虑是否为AS,并及时行基因检测。在一项多中心对36例IgAN患儿进行Ⅳ型胶原蛋白的基因检测的回顾性研究中发现[10],4例(4/36)患儿存在COL4A3变异,未检出COL4A4及COL4A5变异,并认为COL4A3变异是导致IgAN严重发作的易感因素。已知IgA只是一种免疫荧光病理诊断,诊断时要结合临床,已知部分正常人也可以有肾组织IgA沉积,因此,不是所有IgA沉积者均诊断为IgAN。全基因组关联研究(GWAS)将某些血管紧张素(ACE)基因多态性确定为IgAN进展的独立危险因素[11],在IgAN的发病机制中涉及多个基因,包括导致AS的变异基因[12-14]。
总之,本文首次在国内报导1例常染色体隐性遗传性AS误诊、误治为IgA肾病儿童例,提示临床医师凡是临床过程特别是治疗无效的原发性肾小球疾病如IgA肾病等,注意有无合并遗传性肾小球疾病如AS可能,要意识到肾组织IgA沉积不代表一定就是IgA肾病,临床过程一定要相符,特别是治疗反应。凡是对于临床治疗反应欠佳的任何疾病,均应再次审视原发病诊断有无疑问。
猜你喜欢
中国现代医生(2022年19期)2022-11-04
昆明医科大学学报(2022年4期)2022-05-23
昆明医科大学学报(2021年12期)2021-12-30
中国民间疗法(2021年19期)2021-11-20
中国民间疗法(2021年18期)2021-11-02
基层中医药(2021年8期)2021-11-02
中国自行车(2018年8期)2018-09-26
中国继续医学教育(2015年2期)2016-01-06
西南军医(2015年1期)2015-01-22
祝您健康(2000年11期)2000-12-31
