
P掺杂对Ni3Al晶界结合性能的影响
2023-04-29 00:00:00黄仁忠刘雅娟刘丽丽高天附封文江
沈阳师范大学学报(自然科学版) 2023年5期








文章编号:1673-5862(2023)05-0396-06
摘"""要:晶界对于金属和合金材料的力学性能至关重要。晶界本身相对于晶内结合性能差,是导致合金延展性低的一个重要原因。因此,有必要改善其晶界结合性能,进而提高材料的延展性。利用物理冶金及合金化方法可以改善有序金属间化合物的延展性和可加工性。添加合金化元素可以有效地控制晶界的结构,达到提高晶界结合性能的目的。利用第一性原理方法在原子尺度上研究了P掺杂在Ni3AlΣ5(210)晶界中的偏聚行为及晶界结合性能的增强机制。计算的偏聚能表明,相对于体位置,P原子倾向于向晶界偏聚,且向纯Ni的晶界孔隙中由8个Ni原子围成的间隙位置偏聚的趋势最大。计算晶界断裂过程中的格里菲斯功显示P的晶界偏聚可以改善晶界的结合性能。
关"键"词:金属间化合物; 晶界; 第一性原理; 偏聚能
中图分类号:O77+1""""文献标志码:A
doi:10.3969/j.issn.1673-5862.2023.05.003
Effect of P doping on the binding property of Ni3Al grain boundary
HUANG Renzhong, LIU Yajuan, LIU Lili, GAO Tianfu, FENG Wenjiang
(College of Physical Science and Technology, Shenyang Normal University, Shenyang 110034, China)
Abstract:Grain boundaries are vital to the mechanical properties of metals and alloys. The poor binding properties of grain boundaries are responsible for the low ductility of alloys. Therefore, it is necessary to enhance the binding properties of grain boundaries, so as to improve the ductility of materials. The ductility and machinability of ordered intermetallic compounds can be improved by physical metallurgy and alloying. Alloying elements can be effectively used to control the structures and improve the binding properties of grain boundaries.In this work, the first-principles method is used to investigate the segregation behavior and enhancement mechanism of the additive P in the Ni3AlΣ5(210) grain boundary at the atomic scale. The calculated segregation energy shows that the P atoms tend to segregate towards the grain boundary with respect to the bulk position, especially staying at the interstitial sites surrounded by eight Ni atoms in the Ni grain boundary holes. The calculated Griffith work shows that P doping can enhance the binding property of grain boundaries.
Key words:intermetallic compounds; gain boundary; first-principles; segregation energy
L12型有序金属间化合物Ni3Al具有优异的性能,如优良的抗腐蚀和抗高温氧化性能及低密度与高熔点等性质,是一种非常有前景的高温结构材料[1]。然而,室温脆性断裂和低的断裂抗力限制了它的实际应用[2]。
造成不同金属间化合物的脆性的原因各不相同,但通常可分为环境脆性和本征脆性两类。金属间化合物合金的环境脆性是指它在有水分的环境中发生的室温脆性[3]。金属间化合物的本征脆性通常来源于滑移系统数量不足和晶界弱化[4]。通过控制有序合金的晶体结构,即通过宏观合金化将晶体结构从低对称性转变为高对称性,可以大幅度提高这些合金的延展性[5]。Ni3Al的晶界脆性由2个因素引起:一个是内在因素,由于晶界结合力差,晶界本身相对于晶内结合性能弱;另一个是外在因素,即某些杂质的偏析使晶界变脆。硫已经被确定为一种在Ni3Al中强烈偏析到晶界并导致脆化的微量元素[6]。Ni3Al的晶间断裂与晶界的电子结构有关,这点在实验[7-8]和理论[9-10]上已被证实。Muller等[7]认为Ni3Al中晶界固有的弱点源于晶界附近Ni的d键相比于体中杂化变少。
添加合金元素是改善金属间化合物力学性能的一个有效方法。这些不同的添加元素不仅可以加强基体[11],某些元素还可以明显地偏聚至晶界,可显著增强晶间结合力[12]。例如,B向晶界偏聚,可增强晶界结合强度[13],而H和S向晶界的偏聚则对晶界产生不利影响。
本工作将研究P在Σ5(210)Ni3Al晶界中的偏聚行为。通过计算出的偏聚能和电子结构判断P向晶界的偏聚趋势及P掺杂对晶界结合性能的影响。
1"模型和计算方法
在对纯晶界及掺杂的晶界进行结构优化时,平面波截断能均设置为300eV,k网格选为1×7×3。对所有的体系,当总能量收敛到1.0×10-5~eV·atom-1时,每个原子上的作用力小于0.03eV·Å-1,应力偏差小于0.05GPa;原子位移小于1.0×10-3Å时,结构优化停止。在此次计算中,对自旋极化和非自旋极化的计算都进行了测试,对最后的结论几乎没有影响。因此,在接下来的计算与讨论中仅涉及非自旋极化的结果。
2"结果及讨论
2.1"偏聚能
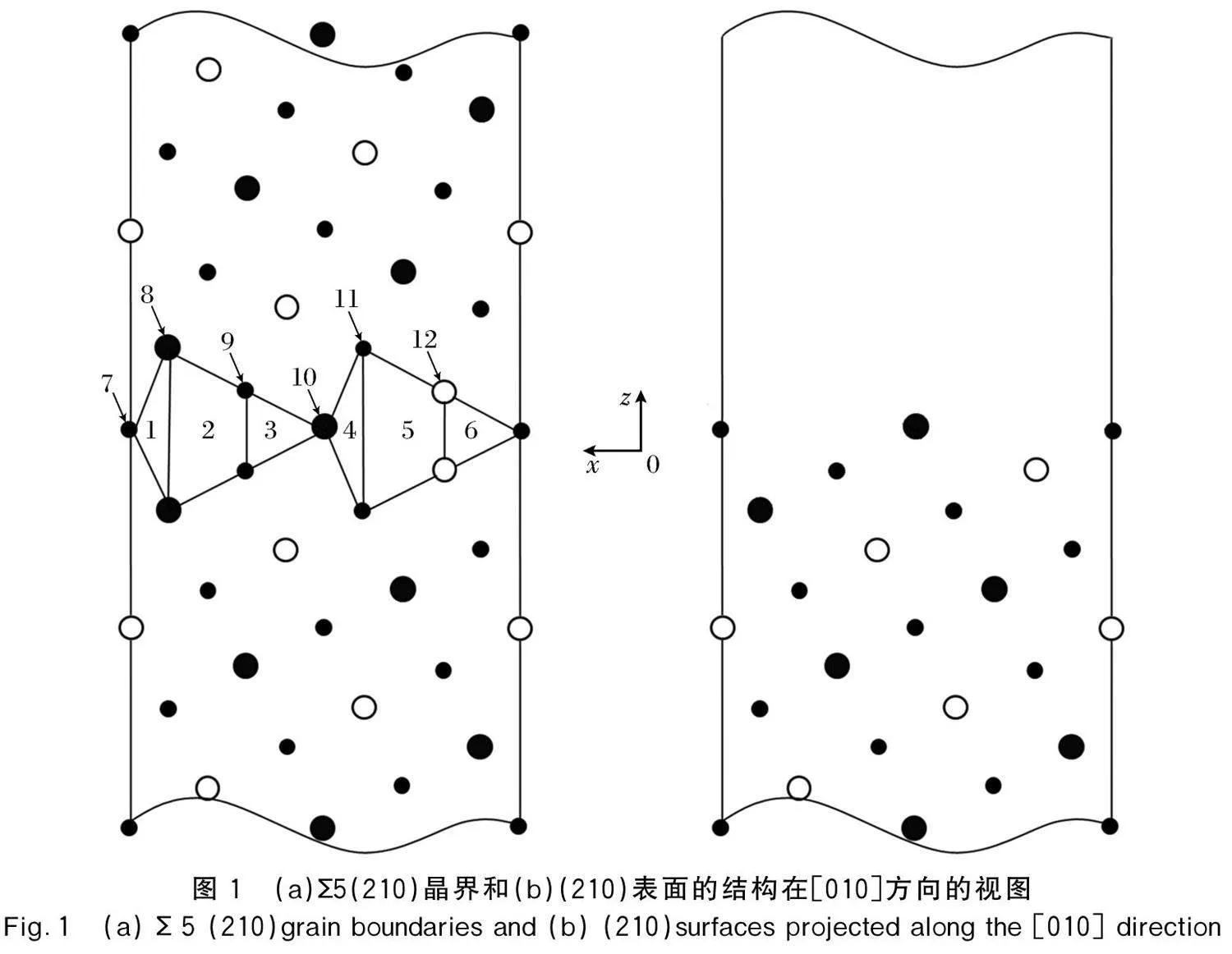
首先,对各种掺P的晶界结构进行优化。弛豫后的结果显示,纯Ni的晶界孔隙中,在I1和 I1′位置的P优化后都不能待在原位置。I1位置的P优化后移动到I2位置处,I1′位置的P优化后移动到I2′位置处;I2和I2′位置的P在优化后都待在原位置;I3位置的P优化后待在原位置,I3′位置的P在优化后移动到I2′位置。为了描述P在晶界结构中的偏聚行为,本文计算了P偏聚到晶界处各种位置的偏聚能。偏聚能定义为有掺杂的晶界或表面的键能与体Ni3Al的键能之差,可以通过公式(1)计算[15]:
混合的晶界孔隙中,I4和I4′位置的P在优化后都待在原位置;I5位置的P在优化后也待在原位置,I5′位置的P在优化后移动到I4′位置;I6位置的P在优化后移动到I5位置,I6′位置的P在优化后仍待在原位置。从偏聚能上来看,I5位置的P偏聚能最负,为-3.681eV·atom-1,这意味着P在混合的晶界孔隙中更倾向于偏聚到I5位置。因此,可以推测,相对于类体位置,P更倾向于向晶界偏聚。
同时,本文还统计了原子的相对位移,即相对于晶界中间层的垂直位移。由于位移的对称性,表2中仅统计了1~10层原子的位移。原子的垂直位移是通过弛豫后的原子位置减去未弛豫前原子的初始位置得到的[17]。结果中正值表示原子远离晶界平面,而负值表示原子靠近晶界平面。未弛豫的晶界结构如图2(a)所示,弛豫后的晶界结构如图2(b)所示。Ni1和Ni2表示位于第2个(210)层的2个Ni原子。
从表2中可以清晰地看到,在没有P掺杂的晶界中,与晶界中间层最近邻的第1层即Ni-Al层上的原子的位移最大,Ni和Al的位移分别为0.319和0.449Å,即Al原子的位移比Ni原子大。这是因为Al原子的键合电荷耗尽,导致Al-Al原子间的静电排斥作用更强[18]。当P位于I2′位置时,使它附近的Ni原子(第1近邻层)与晶界中间层的位移由0.319增加至0.401Å;当P位于I5位置时,使它附近的Al原子的位移由0.449增加至0.566Å。P掺杂的体系原子位置的改变会导致应变能和键能发生变化,进而会影响晶界的结合性能。
除了对12个间隙位置的晶界结构进行分析外,本文也对6种不同替代位置的晶界结构进行优化,计算得到的偏聚能也一并列入表中。在对P替代Ni的5个位置S7~S11位置的偏聚能计算中发现,P替代S9位置的Ni的偏聚能最低为-1.767eV·atom-1,而P替代S12位置的Al的偏聚能为-1.134eV·atom-1,故P在晶界中更倾向于替代Ni位。综上所述,在Ni3Al晶界中两类偏聚位点中,P更倾向于偏聚到间隙I2′位置而不是替代Ni原子。
2.2"分离功
Rice和Wang[19]根据格里菲斯断裂理论[20],详细阐述了晶界断裂过程中的热力学过程与分离功。当P掺杂的晶界被分割成2个表面时,相应的分离功可以用下面的式子来表示:
2.3"差分电荷密度
上述的讨论已经证明P有向晶界偏聚的趋势,且偏聚到晶界可以加强晶界的结合性能。为了揭示晶界强化背后的微观本质,本文研究了P在晶界处的成键特性。以P在I2′间隙位置为例,在未弛豫结构之前,P周围有8个近邻的Ni原子即Ni1~Ni8。弛豫后原子位置发生移动,P-Ni间的距离发生改变。弛豫后P-Ni1之间的距离为2.541Å,P-Ni3之间的距离为2.145Å,P-Ni5及P-Ni6之间的距离为2.254Å,而Ni5-Ni6的原子距离几乎不变。这表明,在图3(a)中的Ni多面体有足够的空间容纳偏析的P,偏析的P的出现几乎没有干扰Ni5-Ni6和Ni7-Ni8的键合。晶界处的Ni—Ni键在GB内聚力中起着至关重要的作用,间隙P的嵌入并没有影响Ni5-Ni6和Ni7-Ni8键的强度即晶界原有的结合强度,而额外的P-Ni键则进一步增强了晶界的结合强度。
为了更直观地看出P掺杂对Ni3Al晶界结合性能的影响,本文绘制了P与周围Ni确定平面上的差分电荷密度图。图3(b)和图3(c)为P位于I2′位置时P与Ni3,Ni4确定的平面和P与Ni5,Ni7确定的平面上的差分电荷密度图。通常来说,原子间电荷重叠越多,所成的键愈强。由图3(b)中可看出,P与最近邻的Ni3,Ni4有明显的电荷重叠,说明P与Ni3,Ni4成键较强。由图3(c)中可看出,P与Ni5和Ni7也有电荷重叠,说明P与Ni5,Ni7也成键。因此,P掺杂偏聚到晶界,可与Ni形成较强的P-Ni键,增强晶界的结合性能,进而抑制Ni3Al晶间断裂[22]。
3"结""语
基于第一性原理计算,本文研究了未掺杂及掺杂P的Σ5(210)Ni3Al晶界的结合性能。计算表明,P对Ni3Al晶界的结合性能具有加强作用。从偏聚能方面看,相对于体结构,P更倾向于向晶界处偏聚。在2个不同的晶界孔隙中,P更倾向于向纯Ni孔隙偏聚,且位于I2′多面体间隙位置时的能量最低。与未掺杂P的晶界相比,掺杂P的晶界,需要更多的分离功才能将体系沿晶界处分离,这表明P的确增强了Ni3Al晶界的内聚性。差分电荷密度图表明,P可以与晶界处的Ni形成较强的P-Ni键,加强了晶间内聚力。因此,可以得出P可向Ni3Al晶界偏聚并强化晶界的结合性能的结论。
参考文献:
[1]MISHIMA Y,OCHIAI S,YODOGAWA M,et al. Mechanical properties of Ni3Al with ternary addition of transition metal elements[J]. Mater Trans JIM, 1986,27(1):41-50.
[2]STOLOFF N S. Physical and mechanical metallurgy of Ni3Al and its alloys[J]. Int Mat Rev, 1989,34(1):153-184.
[3]LIU C T. Environmental embrittlement and grain-boundary fracture in Ni3Al[J]. Scripta Metal Mater, 1992,27(1):25-28.
[4]LIU C T,STIEGLER J O. Ductile ordered intermetallic alloys[J]. Science, 1984,226:636-642.
[5]STOLOFF N S. Ordered alloys for high temperature applications[J]. MRS Proc, 1984,39(1):3-27.
[6]SUN S N,KIOUSSIS N,CIFTAN M. First-principles determination of the effects of boron and sulfur on the ideal cleavage fracture in Ni3Al[J]. Phys Rev B, 1996,54(5):3074-3078.
[7]MULLER D A,SUBRAMANIAN S,BASTON P E,et al. Near atomic scale studies of electronic structure at grain boundaries in Ni3Al[J]. Phys Rev Lett, 1995,75(26):4744-4747.
[8]SUBRAMANIAN S,MULLER D A,SILCOX J,et al. The role of chemistry in controlling the bonding and fracture properties of grain boundaries in L12 intermetallic compounds[J]. Mater Sci Eng A, 1997,239:297-308.
[9]EBERHART M E,VVEDENSKY D D. Localized grain-boundary electronic states and intergranular fracture[J]. Phys Rev Lett, 1987,58(1):61-64.
[10]ALVAREZ J R,REZ P. Calculation of electronic properties of boundaries in Ni3Al[J]. Acta Mater, 2001,49(5):795-802.
[11]MORINAGA M,YUKAWA N,ADACHI H. Alloying effect on the electronic structure of Ni3Al (γ′)[J]. J Phys Soc Jpn, 1984,53(2):653-663.
[12]ZHENG L P,MA Y G,HAN J G,et al. A double species model for study of relaxation of impure Ni3Al grain boundaries[J]. Phys Lett A, 2004,324(2/3):211-218.
[13]MASAHASHI N. Physical and mechanical properties in Ni3Al with and without boron[J]. Mater Sci Eng A, 1997,223(1/2):42-53.
[14]NITYANANDA R,HOHENBERG P,KOHN W,et al. Inhomogeneous electron gas[J]. Resonance, 2017,22(8):809-811.
[15]LOZOVOIA Y,PAXTON A T. Boron in copper: A perfect misfit in the bulk and cohesion enhancer at a grain boundary[J]. Phys Rev B, 2008,77(16):165413.
[16]YAMAGUCHI M,SHIGA M,KABURAKI H. Grain boundary decohesion by impurity segregation in a nickel-sulfur system[J]. Science, 2005,307:393-397.
[17]HU Q M,YANG R,XU D S,et al. Energetics and electronic structure of grain boundaries and surfaces of B-"and H-"doped Ni3Al[J]. Phys Rev B, 2003,67(22):224203.
[18]LU G,KIOUSSIS N,WU R,et al. First-principles studies of the∑5 tilt grain boundary in Ni3Al[J]. Phys Rev B, 1999,59(2):891-898.
[19]RICE J,WANG J S. Embrittlement of interfaces by solute segregation[J]. Mater Sci Eng A, 1989,107:23-40.
[20]GRIFFITH A A. The phenomena of rupture and flow in solids[J]. Philos Trans R Soc Lond A, 1921,221:163-198.
[21]RAZUMOVSKIY V I,RUBAN A V,RAZUMOVSKII I M,et al. The effect of alloying elements on grain boundary and bulk cohesion in aluminum alloys: An ab initio study[J]. Scripta Mater, 2011,65(10):926-929.
[22]JI F,MA S Y,XIN T Z,et al. First-principles study of the effect of boron on grain boundary in NiAl[J]. Comp Mater Sci, 2016,121:1-5.
收稿日期:2023-03-20
基金项目:辽宁省教育厅基本科研项目(LJKMZ20221466)。
作者简介:黄仁忠(1971—),男,河南罗山人,沈阳师范大学教授,博士。
