
固相萃取—气相色谱/质谱法同时测定咖啡干果皮中24种农药残留
2023-03-22 04:11:34梅丽宝尹海飞李树珍龙宇宙
食品与机械 2023年1期
刘 超 梅丽宝 尹海飞 李树珍 龙宇宙
(1.保山市质量技术监督综合检测中心,云南 保山 678000;2.中国热带农业科学院香料饮料研究所,海南 万宁 571533)
咖啡与可可、茶叶并称世界三大饮料,是仅次于石油的第二大世界贸易产品,全球咖啡及其制品的消费人数已超过15亿[1]。作为咖啡副产物的咖啡果皮,质量约占咖啡鲜果的43%~50%,每年中国咖啡生产过程中有40万t鲜果皮产生,除少量用于沤肥外,大量被丢弃[2]。目前,有关咖啡果皮的研究较多,如咖啡果皮成分研究[3-5]、咖啡果皮活性物质分析[6-9]、咖啡果皮茶[10-12]、咖啡果皮酒新产品研发[13-14]等。其中较为成熟的研究是将咖啡鲜果皮晒干后,制成干果皮泡水喝,其味道香甜,深受消费者喜爱。但是,咖啡作为农作物,种植过程中会施用农药,因此咖啡果皮农药残留问题是消费者关注的热点。而有关农药残留的检测方法主要有快速试剂盒法[15]、分光光度法[16]、色谱法[17-18]、质谱法[19-20]等。快速试剂盒法只能初步筛查样品中是否存在农药残留,不能准确定量;分光光度法只适用于部分农药残留的检测;气相色谱法对农药残留假阳性峰不能准确识别,存在误判;气相色谱—质谱法因具有分析准确、灵敏度高、线性范围宽等优点被广泛应用[21-24],已成为当前农残检测技术的重要手段[25-26]。
针对咖啡果皮基质成分复杂:含糖类、脂肪、蛋白质、粗纤维、咖啡因、花青素、绿原酸、葫芦巴碱、生物碱等[27],在农残检测过程中存在基质效应高、回收率低、检测结果不准确的问题。研究拟通过优化提取溶剂、控制洗脱溶剂极性、选择合适的固相萃取净化填料,建立固相萃取净化技术—气相色谱/质谱法同时测定咖啡干果皮中24种农药残留的方法,为咖啡干果皮中多种农药残留的检测提供依据。
1 材料与方法
1.1 材料与仪器
1.1.1 材料与试剂
咖啡干果皮:采自云南省保山市、普洱市、临沧市、德宏州,共12份样品,常温密封贮藏;
农药标准物质(敌敌畏、甲胺磷、异丙威、灭线磷、甲拌磷、二嗪磷、氯唑磷、久效磷、甲基毒死蜱、毒死蜱、甲基对硫磷、倍硫磷、马拉硫磷、杀螟硫磷、对硫磷、二甲戊灵、喹硫磷、水胺硫磷、三唑醇、杀扑磷、溴虫腈、丙环唑、三唑磷、戊唑醇、环氧七氯):质量浓度为100 μg/mL,1.2 mL/支,坛墨质检—标准物质中心;
正己烷、乙腈、丙酮、甲苯:色谱纯,美国Supelco公司;
水:UP超纯水,实验室自制;
ProElut Florisil固相萃取小柱:500 mg/6 mL,迪马科技有限公司;
Mega BE Carb/NH2固相萃取小柱:500 mg/6 mL,美国Agilent Technologies公司;
ProElut TPC固相萃取小柱:3 g/12 mL,迪马科技有限公司。
1.1.2 主要仪器设备
气相色谱—质谱联用仪:7890A-5975C型,配有EI电子轰击离子源,美国Agilent Technologies公司;
石英毛细管色谱柱:ZB-1701型,30 m×0.25 mm×0.25 μm,广州菲罗门科学仪器有限公司;
电子分析天平:MS205DΜ型,瑞士Mettler Toledo公司;
超声仪:AS3120型,北京华瑞博远科技发展有限公司;
氮吹浓缩装置:MTN-2800W型,天津奥特赛恩斯仪器有限公司;
旋转蒸发仪:RE-3000型,上海亚荣生化仪器厂;
离心机:TDL-40B型,上海安亭科学仪器厂。
1.2 方法
1.2.1 混合标准工作溶液的配制
(1)混合标准储备液:以丙酮为溶剂,依次吸取24种农药标准物质各1.00 mL于25.00 mL棕色容量瓶中,定容,摇匀,配制成质量浓度为4.00 μg/mL的储备液。
(2)内标溶液:准确移取质量浓度为100 μg/mL的环氧七氯标准溶液1.00 mL,用丙酮定容至2.00 mL,配成质量浓度为50.00 μg/mL的内标溶液。
(3)混合标准工作液:分别移取0.00,0.20,0.50,1.00,2.50,5.00 mL混合标准储备液于10.00 mL棕色容量瓶中,用空白基质溶液定容,配制成质量浓度分别为0.00,0.08,0.20,0.40,1.00,2.00 μg/mL的标准工作液,移取1.00 mL于1.5 mL进样瓶中,加入20 μL内标溶液,使内标浓度为1.00 μg/mL。
1.2.2 试样的制备 取咖啡干果皮400 g,按四分法取样,粉碎,过0.22 mm孔径筛网。
1.2.3 试样的提取 称取5.00 g试样于50 mL塑料离心管中,加3.0 mL水润湿,加入20 μL内标和30 mL乙腈,盖紧塞子,超声提取30 min,4 000 r/min离心10 min,取上清液,加入20 mL乙腈,重复上述操作,合并离心液于150 mL鸡心瓶中,45 ℃水浴减压蒸发近干,加入2.0 mL丙酮溶解,待净化。
1.2.4 试样的净化 将ProElut TPC固相萃取小柱安装于固相萃取装置上,用5.0 mL乙腈—甲苯(V乙腈∶V甲苯=3∶1)预淋洗小柱,待淋洗液到达吸附层表面时,将浓缩液转移至固相萃取柱中,下接10 mL比色管,用10 mL洗脱液洗脱,控制流速为2~3 mL/min,收集洗脱液于10 mL 比色管中,45 ℃水浴氮吹浓缩近干,用1.0 mL丙酮溶解,定容,过0.22 μm尼龙滤头,供气相色谱—质谱联用仪测定。
1.2.5 分析条件
(1)色谱条件:升温程序为80 ℃保持1 min,以20 ℃/min升至200 ℃,保持7 min,以20 ℃/min升温至265 ℃,保持10 min;载气(He)流速1.0 mL/min,压力70.3 kPa,进样口温度260 ℃,进样量1.0 μL,不分流进样。
(2)质谱条件:电子能量70 eV;传输线温度280 ℃;离子源温度200 ℃;选择离子(SIM)监测模式;扫描时间5~28 min。
1.2.6 方法的定量限、回收率和精密度测定 取咖啡干果皮空白样品,按试验方法处理后,测定被测样品信号强度S与噪音强度N,按S/N=10计算定量限;另取两份咖啡干果皮空白样品,分别在0.03,0.30 mg/kg水平进行加标试验,重复测定6次,计算加标回收率和日内精密度,同时取相同含量的对照品,连续监测6 d,计算方法的日间精密度。
1.2.7 试验分析 依次准确吸取1.0 μL标准工作液及处理后的样品溶液注入气相色谱—质谱联用仪(GC-MS),以保留时间和离子丰度比定性,以峰面积内标法定量。
2 结果与分析
2.1 提取溶剂的选择
查阅相关资料[28-30],农药残留检测常用的提取溶剂有石油醚、丙酮、乙腈。石油醚作为提取溶剂时,主要用于油脂含量高、极性小的有机氯类农药化合物的提取,如GB/T 5009.19—2008《食品中有机氯农药多组分残留量的测定》,对极性较大的农残化合物提取效率低。丙酮作为提取溶剂,适用于大多数农残的提取,特别是极性较大的有机磷类农药的提取,如GB/T 5009.20—2003《食品中有机磷农药残留量的测定》,但丙酮在提取过程中,植物组织中的油脂和色素也会被提取出来,严重影响后续的净化处理[31]。乙腈作提取剂时,提取效率高、溶出杂质少、后处理简便[32]。因此,选择乙腈作为提取溶剂。
2.2 洗脱溶剂和固相萃取小柱的选择
3种不同的洗脱溶剂[正己烷—丙酮(V正己烷∶V丙酮=9∶1)、丙酮、乙腈—甲苯(V乙腈∶V甲苯=3∶1)]对3种不同填料固相萃取小柱(ProElut Florisil固相萃取小柱、Mega BE Carb/NH2固相萃取小柱、ProElut TPC固相萃取小柱)中农残的洗脱效率见表1。
由表1可知,吸附于ProElut Florisil固相萃取小柱中的农药残留洗脱效果为乙腈—甲苯>丙酮>正己烷—丙酮;吸附于Mega BE Carb/NH2固相萃取小柱中的农药残留洗脱效果为乙腈—甲苯>丙酮>正己烷—丙酮;吸附于ProElut TPC固相萃取小柱中的农药残留洗脱效果为乙腈—甲苯>丙酮>正己烷—丙酮,说明选取正己烷—丙酮和丙酮作为洗脱溶剂时,24种农药洗脱效率值均低于乙腈—甲苯溶液,同时误差也最大。当用正己烷—丙酮作洗脱溶剂时,吸附在ProElut Florisil固相萃取小柱和Mega BE Carb/NH2固相萃取小柱上的敌敌畏、甲拌磷较难被洗脱。由图1可知,敌敌畏、甲拌磷在GC-MS中的响应值非常低,主要与农药的结构及洗脱溶剂的极性有关,而被检测的农药多为有机磷类和杂环类结构,极性大,依据相似相溶原理,当用极性较低的溶剂正己烷—丙酮和丙酮时,极性大的农药化合物较难被洗脱,当增大极性,用乙腈—甲苯洗脱时,被检测的农药化合物较易被洗脱。
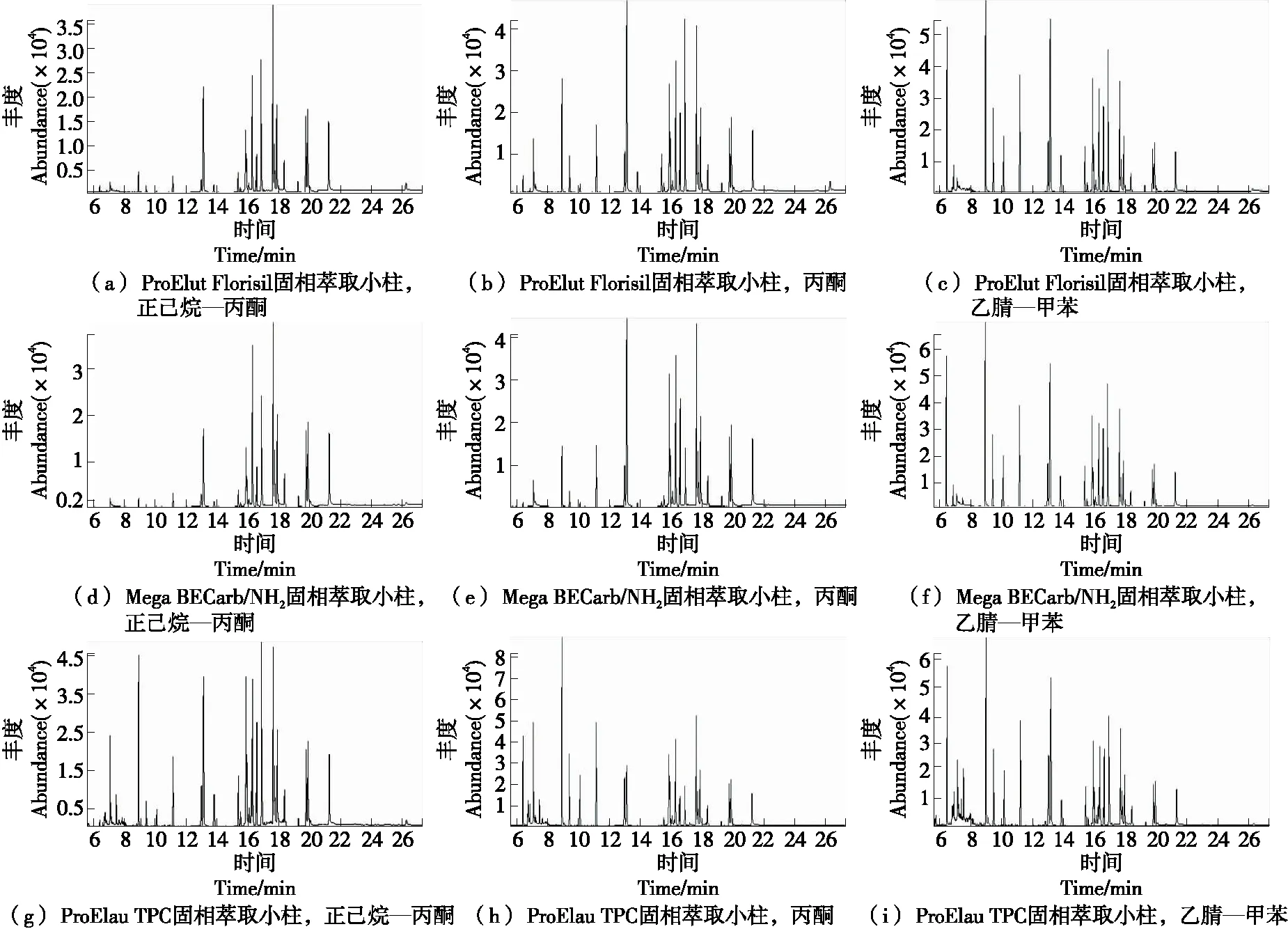
图1 24种农残标准溶液(1.00 μg/mL)在不同固相萃取小柱和洗脱溶剂中选择离子流色谱图

表1 农药标准物质在不同固相萃取小柱和洗脱溶剂下的洗脱效率
在正己烷—丙酮(V正己烷∶V丙酮=9∶1)洗脱溶剂体系下,3种固相萃取小柱的净化效果为ProElut TPC固相萃取小柱>ProElut Florisil固相萃取小柱>Mega BE Carb/NH2固相萃取小柱;在丙酮洗脱溶剂体系下,3种固相萃取小柱的洗脱效果为ProElut TPC固相萃取小柱>ProElut Florisil固相萃取小柱>Mega BE Carb/NH2固相萃取小柱;在乙腈—甲苯(V乙腈∶V甲苯=3∶1)洗脱溶剂体系下,3种固相萃取小柱的洗脱效果为ProElut TPC固相萃取小柱>Mega BE Carb/NH2>固相萃取小柱ProElut Florisil固相萃取小柱。3种固相萃取小柱的洗脱净化效率存在差异,主要与固相萃取小柱的填充材料有关,Florisil固相萃取小柱以无机材料弗罗里硅藻土(MgO·SiO2)为主,是一种高选择性能的吸附剂,主要吸附油脂、烃类含氮化合物及色素;Mega BECarb/NH2固相萃取小柱以石墨化碳(CARB)和氨基(NH2)为主,主要通过离子交换和极性吸附达到保留的作用;ProElut TPC固相萃取小柱是以高聚物PAS、石墨化碳(CARB)和硫酸镁为主,是与NH2相似的吸附柱,但具有比NH2更强的离子交换能力,可用于去除农残分析中的金属离子、有机酸、色素和酚类物质。24种农残在ProElut TPC固相萃取小柱上的吸附效率最低,经GC-MS检测,目标峰最多,其次为ProElut Florisil固相萃取小柱,吸附最大的为Mega BE Carb/NH2固相萃取小柱。
综上,选择乙腈—甲苯(V乙腈∶V甲苯=3∶1)作为洗脱溶剂,ProElut TPC固相萃取小柱作为净化柱。
2.3 分析条件的优化
以两相邻色谱峰分离度≥1.5为原则,优化气相色谱条件,通过全扫描获得混合标准农药总离子流图(TIC),根据总离子流图中目标化合物的保留时间和质荷比(m/z),确定选择离子扫描(SIM)模式下的分析条件,24种农药残留的保留时间、定性离子、定量离子及丰度比见表2。
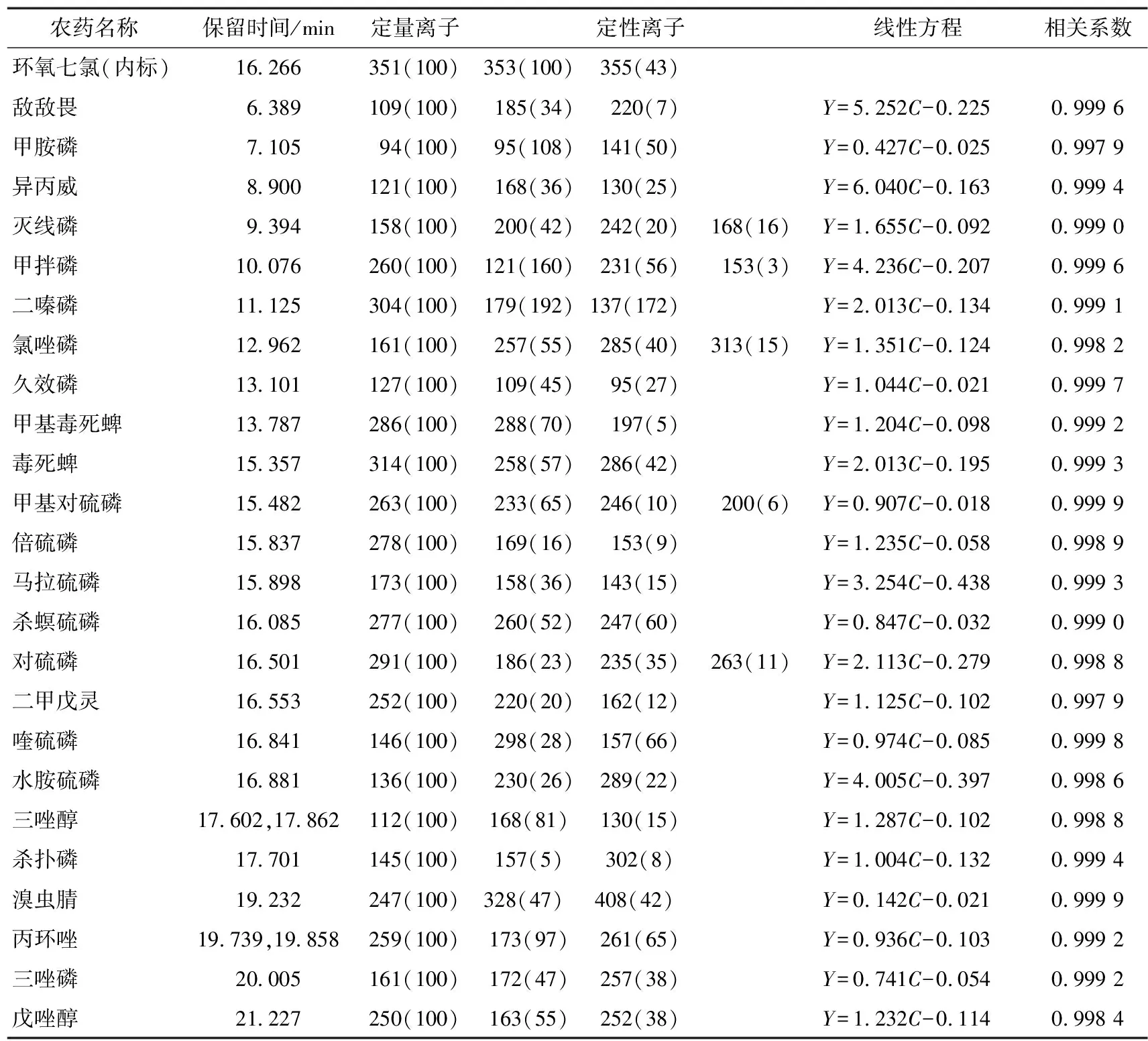
表2 24种农药保留时间、定量和定性离子、线性方程等相关参数表
2.4 校准曲线及其相关性
由图2可知,24种农药在28 min内得到较好分离,各农药在质量浓度为0.016~0.400 mg/kg时线性关系良好,其相关系数为0.997 9~0.999 9,符合GB/T 27417—2017《合格评定 化学分析方法确认和验证指南》定量分析要求。
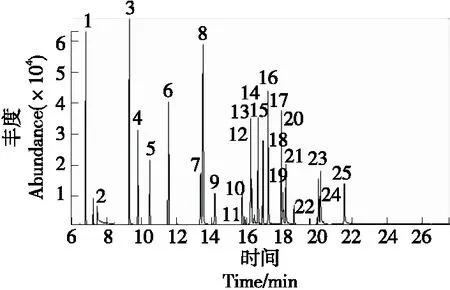
1.敌敌畏 2.甲胺磷 3.异丙威 4.灭线磷 5.甲拌磷 6.二嗪磷 7.氯唑磷 8.久效磷 9.甲基毒死蜱 10.毒死蜱 11.甲基对硫磷 12.倍硫磷 13.马拉硫磷 14.杀螟硫磷 15.环氧七氯(内标) 16.对硫磷 17.二甲戊灵 18.喹硫磷 19.水胺硫磷 20.三唑醇 21.杀扑磷 22.溴虫腈 23.丙环唑 24.三唑磷 25.戊唑醇
2.5 方法的定量限、回收率和精密度
由表3可知,24种农药残留的方法定量限为0.01~0.03 mg/kg;0.03 mg/kg加标水平下,回收率为65.2%~108.5%,日内精密度相对标准偏差(RSD)为3.7%~6.8%,日间精密度相对标准偏差为4.2%~7.2%;0.30 mg/kg 加标水平下,回收率为81.4%~105.7%,日内精密度相对标准偏差为2.5%~5.9%,日间精密度相对标准偏差为2.8%~6.4%,符合GB/T 27417—2017《合格评定 化学分析方法确认和验证指南》要求。

表3 0.03,0.30 mg/kg加标水平下24种农药残留的定量限、回收率和精密度
2.6 实际样品分析
运用建立的方法对保山市、普洱市、临沧市、德宏州4个咖啡主产区的12份咖啡干果皮样品进行农药残留检测,结果表明:2份样品检出毒死蜱,含量分别为0.041,0.037 mg/kg;1份样品检出戊唑醇,含量为0.058 mg/kg,为了进一步验证方法的准确性,将检测出毒死蜱和戊唑醇的样品进液相色谱—质谱/质谱联用仪分析确证,结果一致。依据GB 2763—2021《食品安全国家标准 食品中农药最大残留限量》进行判定,均未超出标准要求;其余样品均未检出,说明云南咖啡果皮农药残留安全状况总体良好。
3 结论
建立了一种同时分析咖啡干果皮中24种农药残留的固相萃取—气相色谱/质谱方法。该方法各性能参数经验证均满足国家标准要求,测定结果准确、可靠,能够运用于咖啡果皮中多组分农药残留的定性和定量分析。当选用乙腈—甲苯(V乙腈∶V甲苯=3∶1)作洗脱溶剂、ProElut TPC固相萃取小柱作净化柱时,净化和洗脱效均最高,效率为95%。从农药残留检出情况看,12批次样品中,3批次样品检出农药残留,依据GB 2763—2021《食品安全国家标准 食品中农药最大残留限量》判定,均未超出标准要求值,表明咖啡干果皮农残安全性较高。同时,由于此次采集的样品数量有限,在农残检测数据充分性上还有待完善,后续将加大对云南各咖啡主产区咖啡果皮农药残留的监测力度,以便更加准确、客观、科学、全面地评价云南咖啡果皮农药残留状况。
猜你喜欢
少年文艺(2022年8期)2022-07-08 10:02:47
中华养生保健(2020年9期)2021-01-18 03:12:36
无机化学学报(2019年2期)2019-02-27 06:53:38
中国经济周刊(2017年6期)2017-03-21 00:59:27
读写算·高年级(2016年3期)2016-05-30 01:53:46
食品界(2016年4期)2016-02-27 07:37:06
大连工业大学学报(2015年4期)2015-12-11 04:06:50
中国医疗美容(2015年1期)2015-07-12 10:06:18
应用化工(2014年4期)2014-08-16 13:23:09
应用化工(2014年7期)2014-08-09 09:20:27
