
婴儿歌舞伎综合征2例报道并文献复习
2023-03-16 02:42:22张坤卢伟能
中国生育健康杂志 2023年2期
张坤 卢伟能
歌舞伎综合征(kabuki syndrom,KS)在1981年由Niikawa和Kuroki两位日本研究者首次报道,也称Niikawa-Kuroki综合征[1-2],日本的患病率约1∶32 000,澳大利亚和新西兰约1/86 000,中国暂无相关统计。KS主要临床表现为特殊面容、智力发育落后、骨骼畸形、皮纹异常、脏器畸形。病因尚不完全清楚,部分有细胞遗传学异常,2010年及2012年先后证实KMT2D、KDM6A基因突变与该综合征有关,称KS-1型和KS-2型[3-4]。本病预后不良,目前无特异治疗,以对症治疗为主,国内大部分KS患者均在临床表型明显出现后才进行分子诊断确诊,此时患儿多已出现严重智力发育落后、生长发育迟缓、听力受损等症状,因此婴儿早期,甚至在新生儿期识别KS至关重要,早期干预可能有望改善预后。本文报道2例婴儿KS,结合相关文献,对国内目前在婴儿期诊断KS相关病例报道进行归纳总结,了解KS婴儿早期主要表型及临床特点,提高临床医生认识。
资料与方法
1.临床资料:病例1,男,37+6周,顺产出生,出生体重3 940 g,阿氏评分正常,父母及9岁姐姐体健,母亲规律产检,OGTT阴性,孕期血糖正常,唐氏筛查低风险,产前提示胎儿迷走右锁骨下动脉,胎儿左右心比例失调,卵圆瓣冗长,行羊水穿刺查染色体微阵列无异常。生后母婴同室,不久监测血糖低至1.1 mmol/L,收入院。查体发际线低,耳位低,双手通贯掌,心肺腹查体无异常。合并继发性呼吸窘迫,无创辅助通气支持5 d,生后2d总胆红素高至275 mmol/L,ABO血型不合相关性溶血,予蓝光照射、人免疫球蛋白治疗。早期持续性低血糖,需静脉输注葡萄糖维持血糖正常,肝功能、血氨基酸、酰基肉碱、尿有机酸检查无异常,甲功三项、皮质醇、生长激素、促肾上腺皮质激素无分泌低下,在血糖1.7 mmol/L时行胰岛素(4.38 uIU/mL)、C-肽(1.2ng/mL)检测,排除早发败血症、低体温、红细胞增多症、喂养不足等情况后,考虑暂时性高胰岛素血症,生后一周合并感染,血培养、脑脊液培养提示肺炎克雷伯杆菌(耐药菌),经美罗培南抗感染治疗3周、2次血培养脑脊液培养阴性后停药。头颅核磁共振及心脏彩超检查无异常。期间患儿存在喂养困难,吸吮力量欠佳,吞咽尚可,进食缓慢,耐力下降。加强吸吮吞咽训练,分次多餐喂养,耐受后出院,2月龄基因确诊,3月龄随诊Alberta婴儿运动量表得分为4;百分位<5TH。4月龄Bayley评估:粗大运动低于平常,认知非常落后,余均轻度落后。定期康复治疗。
病例2,男,36+1周,胎膜早破顺产出生,出生体重2 250 g,母亲规律产检,无特殊异常,父母及3个姐姐体健。查体耳位低,左耳前瘘管,右耳前外侧2个米粒大小赘生物,生后不久出现呼吸困难,2.5号气管插管声门下有阻力,生后11 d转入本院。行颌面部CT+三维重建示双侧后鼻孔膜性闭锁、先天性气道狭窄,心脏彩超动脉导管未闭(4 mm)卵圆孔未闭(2.9 mm)、轻度肺动脉高压,头颅超声未见异常,鼻饲喂养有反复溢奶、呕奶。家人由于个人原因于患儿生后20 d放弃治疗,死亡18 d后基因回报确诊。
2.方法:分别取2例患儿及其父母外周全血采用高通量测序技术对 4 811个单基因遗传病基因进行突变筛查,并对检测结果进行生物信息学分析。在PubMed数据库中,分别输入“Kabuki syndrome”“Niikawa-Kuroki”“KMT2D”“KDM6A”等检索文献。输入“歌舞伎综合征”“kabuki综合征”等在万方数据库、中国知网数据库检索文献。检索时间设为2010年8月—2021年3月,纳入文献标准为(1)报道病例为中国患儿;(2)报道病例在1岁内诊断KS综合征;(3)存在KMT2D或KDM6A基因突变。最后归纳总结。
结果
1.基因检测结果:病例1为KMT2D,NM_003482.3;c.1583delC(p.Pro528HisfsTer402) ,无义突变,在 dbSNP、1 000 Genomes 及 gnomAD 数据库未见收录。生物信息学软件预测该突变会导致蛋白质截断,提前出现氨基酸终止密码。病例2为KMT2D,NM_003482.3;c.10775T>C变异(p.Met3592Arg),错义突变,同样未见收录。生物信息学软件预测该突变会破坏野生型蛋白质结构/功能。2例均根据 Clingen 数据库,KMT2D基因为单倍剂量不足敏感基因(Haploinsufficiency Score:3)。综合家系分析,父母未检出该突变,均为新发突变(不排除父母存在生殖腺或体细胞嵌合可能)。突变临床分级为致病性。
2.病例统计分析:共检索到14篇符合条件文章,其中英文7篇[5-11],中文7篇[12-18],结合本文报道的上述2例患儿,共收集29例患儿临床资料,中位诊断年龄3月,3月龄内18例,3~6月龄5例,6~12月龄5例,另外1例26月龄基因诊断,因婴儿期资料详细,也纳入统计。男21例,女8例。21例(72.4%,21/29)生后于新生儿科住院诊治,呼吸困难9例(31.0%,9/29),喂养困难17例(58.6%,17/29),颅内结构发育异常3例(10.3%,3/29),心脏发育异常15例(51.7%,15/29),后鼻孔闭锁1例(3.4%,1/29),消化道畸形3例(10.3%,3/29),泌尿生殖系统畸形5例(17.2%,5/29),听力异常11例(37.9%,11/29),败血症5例(17.2%,5/29),新生儿低血糖10例(34.4%,10/29),其中1例加用激素、1例加用二氮嗪,肌张力低13例(44.8%,13/29),甲状腺功能减低3例(10.3%,3/29)。27例(93.1%,27/29)存在特殊面容描述,2例(6.9%,2/29)面部特征不详按正常统计,长眼裂和/或下眼睑外翻65.5%(19/29),拱形眉、外侧眉毛稀疏缺失41.3%(12/29),鼻尖凹陷、鼻柱宽55.1%(16/29),大耳、耳位低、耳前小凹65.5%(19/29),腭裂、高腭弓58.6%(17/29)。小下颌24.1%(7/29),贯通掌24.1%(7/29),第五小指发育不良、胎脂垫41.3%(12/29)。见表1。
本文29个患儿中KMT2D基因相关变异25个,从第4外显子到第53外显子分布不均,9个位于第39外显子,3例具体突变位点不详,其中1例为试管婴儿孕中期查染色体核型46,XY,t(6;12)q25:q24为已报道的KS相关染色体变异。4例为KDM6A基因突变,其中2例分别位于4号和18号外显子,另外2例不详,2例移码突变。详见表2。
讨论
关于KS的诊断主要依据典型的临床表现,结合分子诊断技术证实。早期面容不典型,难以识别,诊断会被延误,需根据其他临床表现分析做出诊断。国外早期的一些回顾性分析研究中几乎没有在婴儿期诊断的KS,近年来,随着人们对该疾病的认识增加和分子诊断技术的发展,婴儿期诊断KS的病例逐渐增加。Dentici[19]等研究显示,婴儿期通过临床诊断KS中位年龄为3.6月龄。本文29例患儿诊断的中位年龄为3月龄,报道2例结合基因检查在生后1~2月内确诊,基因检查可能帮助更早诊断KS。
本文29例患儿中2例无面容相关介绍按正常统计,长眼裂和/或下眼睑外翻约65.5%,拱形眉、外侧眉毛稀疏缺失新生儿期未描述,41.3%婴儿期被观察到。55.1%病例有鼻尖凹陷、鼻柱宽记录。大耳、耳位低、耳前小凹为65.5%,国外病例几乎均见到耳朵外观异常[19];2个病例有发际线低描述,比较少见。腭裂、高腭弓发生率稍高为58.6%,可能该异常易观察;24.1%有小下颌,是KS患者口腔异常表现之一;在KS患儿中通贯掌并无特异性,国外未见相关统计,该统计中27.5%患儿存在;第五小指发育不良和胎指垫是KS,是骨骼畸形和皮纹异常的具体表现,仅41.3%病例被描述,低于国外相关统计(87%)[19],这方面的认识还需加强。
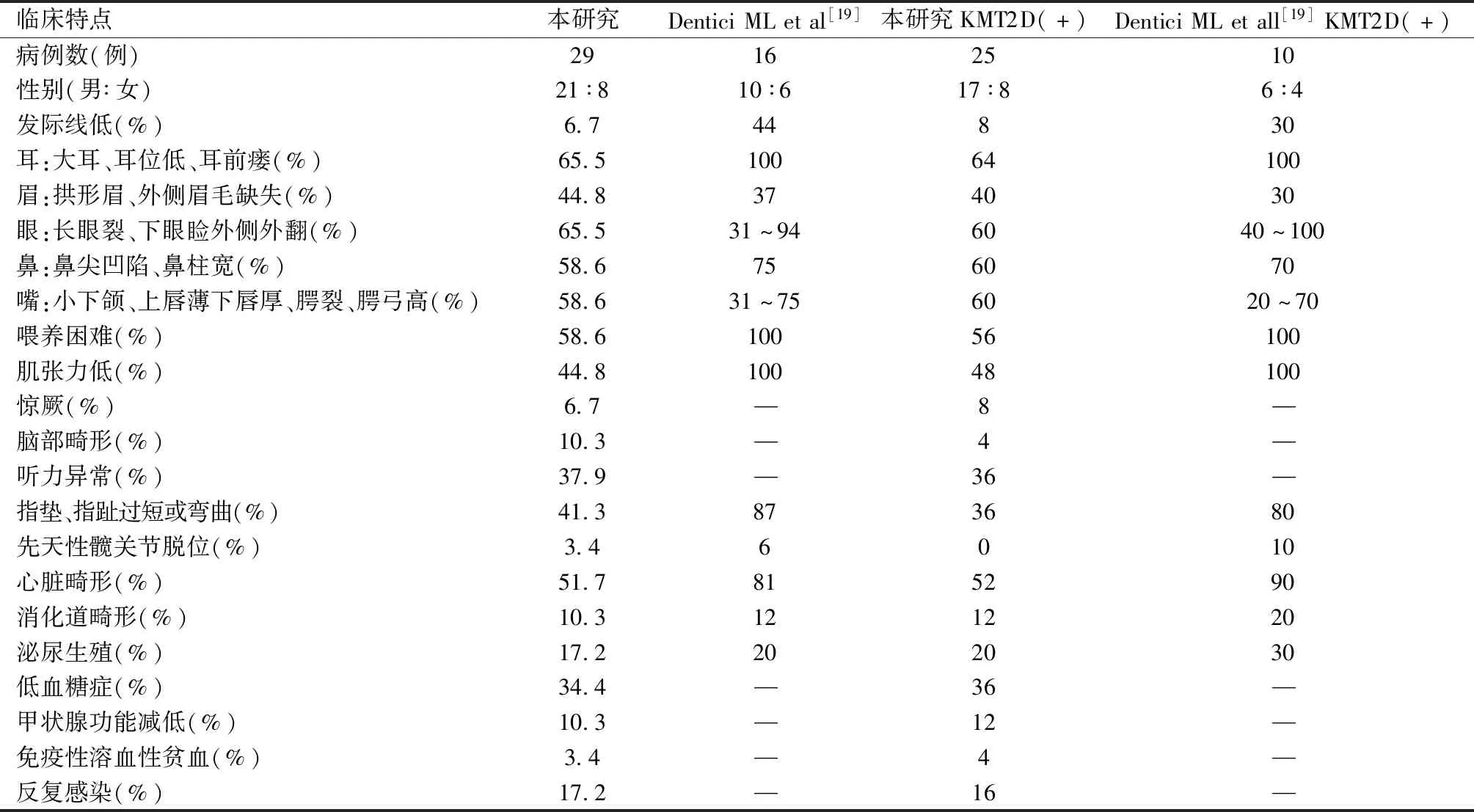
表1 29例患儿临床表型情况
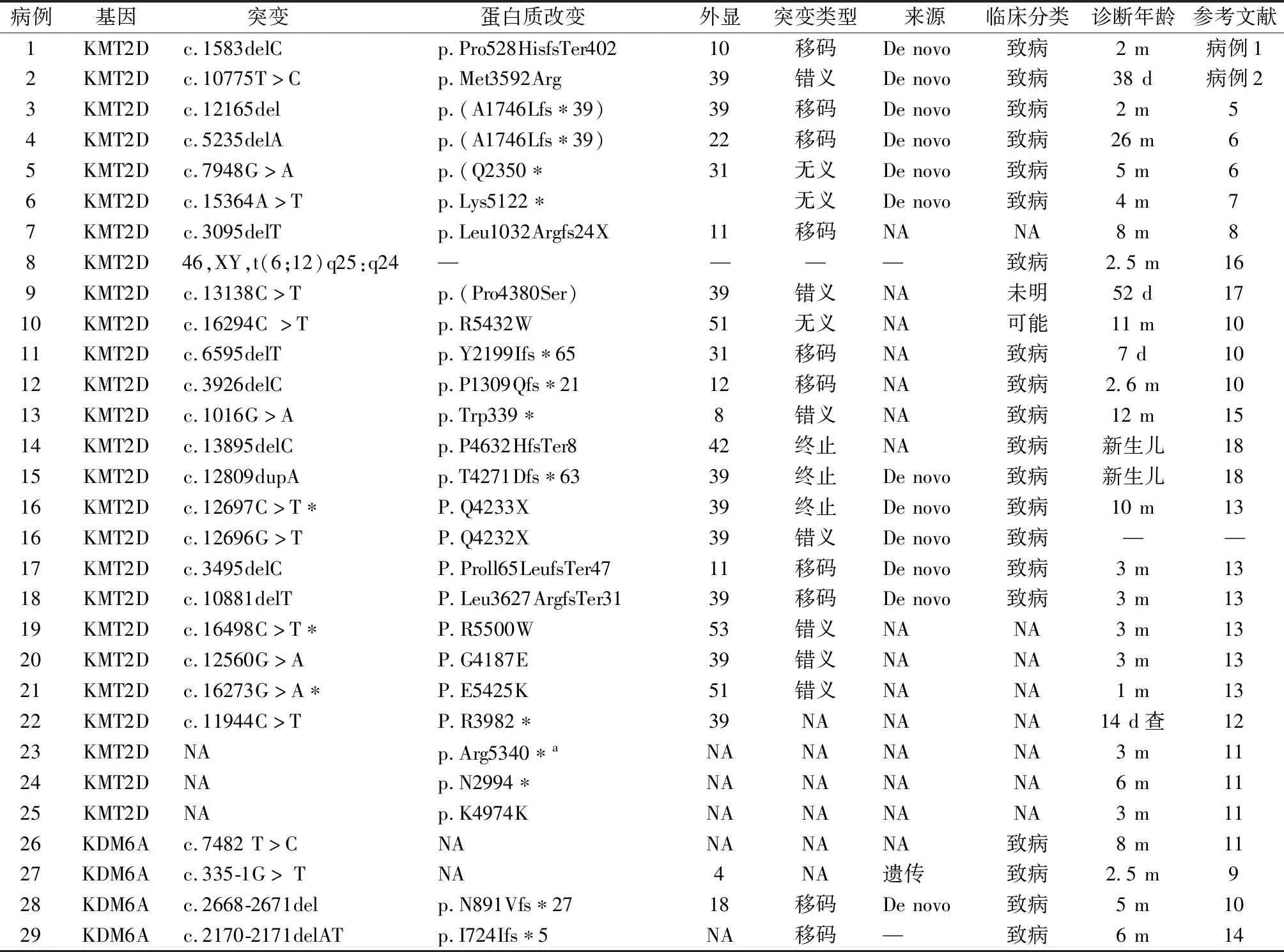
表2 29例患儿基因异常情况
本文在脏器缺陷中发现心脏发育异常约51.7%,包括室间隔缺损、房间隔缺损、动脉导管未闭、主动脉缩窄发育不良,与国内KS先心病方面的统计相似42.6%[10],较国外统计偏低[19]。泌尿生殖系统畸形约17.2%,包括异位肾、肾囊肿、多囊肾、隐睾、输尿管扩张,与Dentici等[19]统计的发生率接近,国外部分病例中可见马蹄肾、重复集合系统畸形[19]。本文颅内结构发育异常3例,包括Dandy-Walker畸形[5]、小脑蚓部发育不良[6]、先天性脑积水[9]各1例。本文病例中消化道畸形少见,2例肛门闭锁及1例中肠旋转不良,目前未见具体发生率统计,仅为个案报道。
功能异常方面喂养困难、肌张力低被认为是新生儿KS综合征临床特点。Dentici等[19]统计的16例婴儿期诊断KS无论有无基因异常均存在喂养困难、肌张力低。本文统计发生率分别为58.6%、44.8%,部分吸吮吞咽不协调的患儿需要放置鼻胃管,甚至行胃造口术喂养。目前KS约6%~8%会在新生儿期发生低血糖,高胰岛素血症、生长激素缺乏、垂体肾上腺功能不全可能是发生低血糖症的原因,具体机制尚不明确,可能占先天性高胰岛素血症的1%[20]。本病例34.4%存在新生儿低血糖,大部分通过输注葡萄糖维持逐渐过渡,个别加用二氮嗪、激素治疗。听力异常在婴儿期很难通过临床发现,主要依赖脑干听觉诱发电位等检查识别,本文病例37.9%存在听力异常,而邱士伟等[21]分析462例KS患儿中101例听力下降,其中30例存在反复中耳炎,75%听力下降类型不明。
KMT2D和KDM6A是引起KS的两个已知基因,分别约占75%和3%~5%[22]。KMT2D位于12号常染色体显性遗传,含54个外显子[23],本文29个患儿中KMT2D基因相关变异25个,9个位于第39外显子,符合Bogershausen等[23]的研究,该研究认为其是热点突变。KDM6A基因位于Xp11.3染色体上,为X连锁遗传,包含29个外显子[23],本文共统计4例,其中2例分别位于4号和18号外显子,另外2例不详。统计的29例基因突变中大部分是无意义和移码突变,导致蛋白质明显改变致病。某些错义突变导致氨基酸替换不一定会损害蛋白质功能,需选择合适的工具谨慎判断致病性[22]。
总之,婴儿期与KS相关的临床特征谱广泛,面部表型在婴儿期不太明显,如拱宽眉形眉毛等。对于一些临床特征,如大耳朵和长眼睑裂缝[19],可以提示临床怀疑KS,特别是合并低血糖、肌张力低、喂养困难、畸形时,分子检查可帮助确诊。目前,尽管KS尚不能被治愈,但早期诊断对于前瞻性治疗相关的医学问题和遗传咨询至关重要,有望改善预后,本文报道的病例1目前定期于康复科治疗干预中。
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29 01:57:52
疯狂英语·新悦读(2021年1期)2021-01-27 10:43:00
中国生殖健康(2020年4期)2021-01-18 02:58:10
科学生活(2019年7期)2020-01-01 08:28:02
养殖与饲料(2019年10期)2019-02-25 14:52:37
中国生殖健康(2018年4期)2018-11-06 07:12:16
山东畜牧兽医(2018年3期)2018-04-26 09:10:34
环球时报(2017-06-27)2017-06-27 19:42:54
河南外科学杂志(2016年2期)2016-03-08 09:34:27
听力学及言语疾病杂志(2015年5期)2015-12-24 01:47:04
