
氰化尾渣矿浆电解液中铁离子的萃取富集
2023-03-01 07:40包进宋永辉董萍李一凡朱荣燕廖龙
化工进展 2023年1期
包进,宋永辉,董萍,李一凡,朱荣燕,廖龙
(1 西安建筑科技大学冶金工程学院,陕西 西安 710055;2 陕西省资源与黄金重点实验室,陕西 西安 710055)
目前,氰化法仍是黄金冶炼行业最普遍的提金工艺,但其在生产过程中会带来环境污染隐患。据统计,我国每年产生的氰化尾渣数量约有2.45×107t,累计积存量超过1.5×109t[1−2]。氰化尾渣中含有氰化物和重金属,被列入危险固废。因此,氰化尾渣的无害化、减量化、资源化是黄金冶炼行业可持续发展面临的关键问题。现阶段氰化尾渣有价金属的综合回收主要有氧化−浸出法、磁化焙烧、高温氯化焙烧、熔融氯化挥发法、浮选法等,但其过程长且复杂[1−3]。西安建筑科技大学宋永辉等[4−5]提出了采用矿浆电解法无害化处理氰化尾渣的技术,可同时实现氰化物的阳极氧化、重金属的阳极浸出与阴极还原沉积,具有工艺简单、流程短、能耗低等优点,有很好的潜在应用价值。矿浆电解过程中,氰化尾渣中大量的黄铁矿也会被氧化,铁以离子形式进入溶液中[6],这为回收氰化尾渣中的铁奠定了基础。
溶液体系中铁的回收与利用的方法有离子交换树脂[7−8]、沉淀法[9]和溶剂萃取法[10−11]等。离子交换树脂传质速率低、装载能力有限,适用于处理低浓度含铁溶液。而沉淀法药剂添加量大,工艺复杂,处理后的废水不达标,对土壤和地下水造成严重污染[12]。溶剂萃取法具有处理容量大、可有效分离杂质及高回收率等特点,是一种有效回收溶液中铁离子的方法[13]。目前,研究较多的萃取剂包括胺类萃取剂N235 及酸性磷(膦)类萃取剂P204、P227 和P507,前者主要在酸性氯介质中提取铁,通过离子缔合提取氯阴离子[14],后者主要用于从酸性溶液中提取含铁的阳离子[15−16]。Wang 等[17]研究表明,N235对Fe3+的萃取效率高,但其稀释时易形成溶解度较小的聚合物,出现不利于萃取的第三相。此外,多种酸混合介质中铁离子与胺提取剂的离子缔合能力较弱,不能达到理想的除铁效果。张晓峰等[18]研究了盐酸体系中P227 萃取铁,虽然铁的萃取率最高能达到99%,但其对萃取体系酸度要求高,pH 高时易形成严重的乳化现象。Wang 等[19]用P507 溶剂萃取硫酸浸出液中的铁,最佳条件下可去除97.6%以上的Fe(Ⅲ),但反萃比较困难。P204作为另一种酸性萃取剂,萃取Fe3+具有分相快速、无第三相、萃取温度范围广等优点。Hu 等[20]用P204从高浓度盐酸溶液中提取Fe3+,阐述了其萃取机理并证明了所提取物种为FeClA2·4HA。采用P204 萃取铁虽然已有许多研究报道,但大多偏重于从其他溶液中去除杂质铁离子[20−21],一般浓度不是很大,很少有将铁直接萃取富集制备主要产品的研究。本文在矿浆电解无害化处理氰化尾渣的同时,采用P204−磺化煤油萃取体系从高硫酸氰化尾渣矿浆电解液中富集回收铁离子,重点研究了各因素对Fe3+萃取率的影响及SO2-4存在时的萃取过程,并采用草酸反萃负载有机相,得到分离纯化后的含铁络合物,为后续光催化实现从氰化尾渣危险固废中得到草酸亚铁产品提供可行性,本工艺为氰化尾渣无害化、资源化处理工艺的完善奠定了基础。
1 实验部分
1.1 实验原料
陕西某金矿的氰化尾渣在常温、NaCl 添加量1mol/L、氰化尾渣100g/L、电流1.5A、电解时间5h的条件下进行矿浆电解处理,处理后尾渣中游离氰和金属氰络离子被氧化去除,同时黄铁矿被氧化后铁离子进入电解液[4],电解液的主成分分析结果如表1所示。

表1 氰化尾渣矿浆电解液多元素浓度分析结果单位:mg/L
1.2 实验步骤
图1 为氰化尾渣无害化处理主体工艺流程图。氰化尾渣由矿浆泵输送到电解槽中进行矿浆电解,固液分离后溶液采用P204−磺化煤油萃取体系萃取铁,负载有机相采用草酸溶液进行,反萃后有机相返回萃取环节循环利用,反萃液进一步处理得到草酸亚铁产品。将P204 与磺化煤油均匀混合得到P204 体积分数为25%的有机相,在25℃条件下将有机相与电解液按照相比1∶1充分混合,固定振荡时间10min 及振荡频率180r/min,随后移入梨形分液漏斗中静置,待分层后分离有机相和水相并测定两相中铁离子含量。将1.0mol/L草酸溶液与负载有机相按相比1∶1充分混合,在35℃条件下水浴振荡10min后移入分液漏斗,待静置分层后分离有机相与水相,测定反萃液中铁离子含量并计算反萃率。
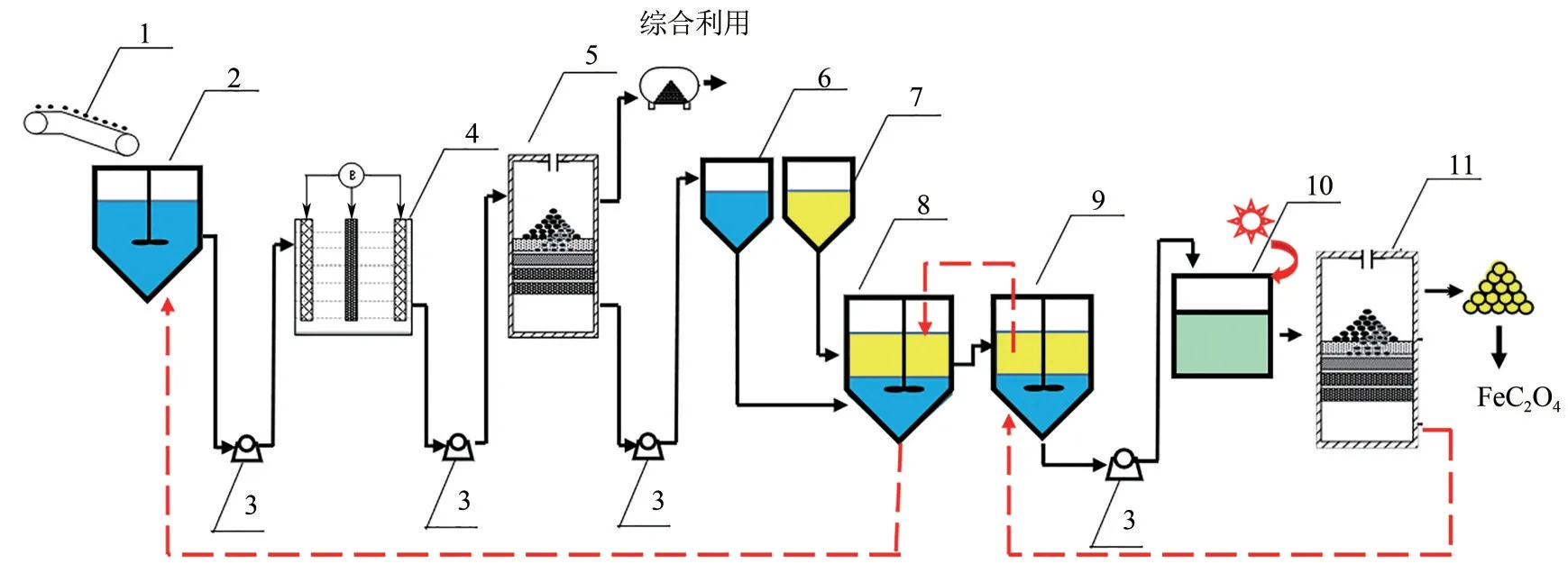
图1 氰化尾渣中铁的回收工艺流程图
1.3 分析表征
有机相的分析表征采用Nicolet Summit 型红外光谱仪与Thermo SClentific Q Exactive组合型四极杆Orbitrap 质谱仪;用火焰原子吸收光谱分析水相中Cu、Fe、Zn等离子浓度,按式(1)~式(3)计算萃取率E、分配比D及选择性分离系数β。
式中,V0、Vaq和Vorg分别表示萃取前水相、萃取后水相和有机相体积,mL;[M]0、[M]aq和[M]org分别表示萃取前水相、萃取后水相和萃取后有机相中金属离子浓度,mg/L;D(M1)和D(M2)分别为金属离子M1和M2的分配比。
2 结果与讨论
2.1 萃取工艺
2.1.1 电解液中多离子的萃取分离
由表1可知,矿浆电解得到的电解液中除含有Fe3+以外,还有部分Fe2+及Cu2+、Zn2+、Na+等存在,因此必须考虑萃取过程中各离子的分离效果。在常温条件下,取初始pH为1.5,固定相比O/A为1∶1,振荡时间5min,振荡频率140r/min,P204 体积分数10%进行萃取实验,结果如图2 所示。Fe3+的萃取率为68.17%,远高于其他离子。由式(3)可计算出P204对Fe3+/Zn2+、Fe3+/Cu2+、Fe3+/Fe2+及Fe3+/Na+的分离系数β分别为47.87、736.89、950.49 及3294.91,说明P204对Fe3+具有很高的选择性。另外,溶液中金属离子的萃取顺序往往取决于金属离子配合物的稳定性常数,这与电子的稳定性、离子电荷、轨道能量以及离子半径等有关[22],Fe3+比Cu2+、Zn2+及Na+具有更高的离子电荷,同时Fe3+具有更高的氧化性,更容易形成配合物。因此,该体系中的几种金属离子萃取顺序为Fe3+>Zn2+>Cu2+>Fe2+>Na+,这也为铁的有效分离和富集奠定了基础。

图2 铁及其他金属离子的萃取率
2.1.2 P204体积浓度对萃取率的影响
为了进一步提高铁的富集回收率,采用浓度为10g/L 的过硫酸铵作为氧化剂,将电解液中的浓度为334.21mg/L的Fe2+氧化为Fe3+,氧化后溶液中Fe3+浓度可达到3682.8mg/L,反应如式(4)所示。改变P204体积浓度进行萃取实验,结果如图3所示。可以看出,随着P204体积分数增大,Fe3+萃取率逐渐增大,25%时达到最大值90.63%,随后不再发生明显变化。萃取剂浓度增大有利于萃取反应正向进行,但P204体积分数过大会造成有机相黏度增大,不利于萃取过程的进行,同时也会增加萃取成本。

图3 P204体积分数对萃取率的影响
2.1.3 电解液pH对萃取率的影响
电解液初始pH为1.5,为探索pH对萃取率的影响,取P204体积分数为25%,调整pH在0.7~2.5进行实验,结果如图4(a)所示。Fe3+萃取率随pH的增大呈现出先增加后平缓的趋势,pH为1.5时达到最大值90.63%,有机相负载Fe3+浓度达到3337.7mg/L。由萃取反应式(7)可知,溶液酸性越低,越不利于反应的正向进行。此外,Fe3+萃取率E与其分配比D呈正相关,而D受P204浓度与pH的影响很大,pH越高D值也就越大[23−24],但pH升高到一定程度,Fe3+会发生水解,因此最大萃取率是在接近Fe3+水解的pH 处。在pH 范 围0~5.5、Fe3+浓 度3682.8mg/L、Cu2+浓度5.63mg/L、Zn2+浓度32.45mg/L、SO2−4浓度10700mg/L、Cl-浓度484.3mg/L 的条件下,采用Visual MINTEQ 软件[24]计算出铁、铜、锌的化学溶解平衡曲线,如图4(b)所示,氰化尾渣电解液中铜、锌以游离Cu2+、Zn2+形式存在。在pH<1.5 时,溶液中Fe 主要以Fe3+、FeSO4+和极少量FeCl2+的形式[式(5)]存在;在pH>1.5 后,溶液发生水解反应[式(6)、式(7)]产生Fe(OH)2+,Fe(OH)2+进一步转变为Fe(OH)3沉淀,导致电解液中铁总浓度降低,此时的萃取率不变,这使得负载有机相中Fe3+浓度降低,这对萃取回收富集电解液中的铁不利,同时也印证了上述pH 越大,负载有机相中Fe3+浓度越低的观点。值得注意的是,pH>1.5时电解液中铁的萃取率依旧能稳定在90.96%,说明电解液中游离Fe3+和FeSO4+均能被P204萃取。
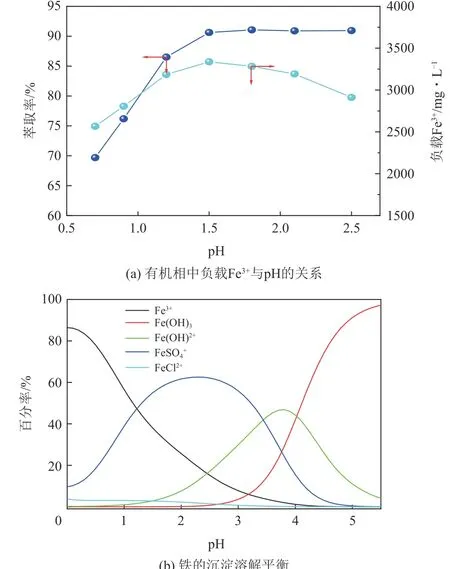
图4 pH对萃取率的影响
2.1.4 O/A对萃取率的影响
在P204 体积分数25%、pH 为1.5 的条件下,考察了相比(O/A)在0.4~2.0 范围内对Fe3+萃取率的影响,结果如图5 所示。Fe3+萃取率随O/A 的增大逐渐增加,在O/A=1∶1 时达到90.72%,此时有机相中Fe3+浓度为3.34g/L。O/A 继续增大,萃取率曲线趋于平缓,但有机相中负载的Fe3+浓度降低。O/A 过大会导致有机相中Fe3+浓度降低,富集效果变差;太小则会有大部分Fe3+保留于水相中,同时会产生乳化现象,分相困难。

图5 O/A对萃取率的影响
2.1.5 振荡时间及振荡频率对萃取率的影响
在P204 体积分数为25%、pH 为1.5、O/A=1∶1的条件下,振荡时间与振荡频率对铁萃取的影响实验结果如图6所示。控制振荡时间为7min,铁的萃取率随振荡频率的增大而增大,180r/min时可达到96.96%(曲线a)。控制振荡频率为180r/min,铁的萃取率随着振荡时间增加而增大,10min时即可达到最大值97.73%(曲线b)。振荡时间与振荡频率是影响有机相与电解液之间传质混合程度的重要因素,增加振荡时间与振荡频率,水相中的Fe3+通过相际界面的速度加快,有机相和水相混合更充分,Fe3+的萃取率增大。

图6 振荡时间及振荡频率对萃取率的影响
2.1.6 萃取温度对萃取率的影响
在P204体积分数为25%、pH为1.5、O/A=1∶1、振荡时间10min 和振荡频率180r/min 的条件下,温度对萃取过程的影响结果如图7 所示。可以看出,铁的萃取率随温度的升高逐渐增大,溶液黏度随温度的升高逐渐减小,Fe3+通过相际界面由水相进入有机相的速度加快,分相时间减小,有利于Fe3+的萃取。进一步提高反应温度,Fe3+萃取率的增加不是很明显,考虑到经济因素,确定萃取温度为25℃。

图7 温度对萃取的影响
2.1.7 饱和萃取容量测定
在P204 体积分数为25%、振荡时间10min、O/A=1∶1、振荡频率180r/min、温度25℃的条件下,取5mL P204、15mL 磺化煤油,每次取20mL 矿浆电解液,考察P204萃取Fe3+的饱和容量,结果如图8 所示。Fe3+萃取率随萃取次数的增加呈现出逐渐降低的趋势,有机相中负载Fe3+浓度呈现先增加后平缓的趋势,这是因为随着萃取次数的增加,未参与萃取反应的P204 浓度逐渐减小,同时,有机配合物浓度的升高抑制了萃取反应的正向进行。经过9次萃取后,P204负载铁容量达到饱和值21.57g/L,Fe3+由水相转移到有机相并富集了5.86 倍,说明此萃取方法能够有效实现Fe3+富集。

图8 P204饱和萃取容量
2.1.8 平行实验
在P204体积分数为25%、O/A=1∶1、振荡时间10min、振荡频率180r/min、温度25℃的条件下进行平行实验,结果如表2 所示。Fe3+的平均萃取率为97.70%,负载有机相中Cu2+和Zn2+平均浓度分别为0.067mg/L 和3.5mg/L,可以看出用P204 萃取电解液中Fe3+是可行的,萃取率高且稳定,同时能有效分离Cu2+、Zn2+等杂质。

表2 P204萃取平行实验
2.2 萃取过程分析
2.2.1 Fe3+萃取平衡及化学计量学
P204 属于磷酸酯类萃取剂,通过螺旋间的氢键以二聚体形式存在[25],Fe3+和FeSO4+萃取平衡主要由P204 二聚体控制,释放的H+转移到水相,而Fe3+和FeSO4+则以配合物的形式从水相进入有机相。其萃取反应如式(8)、式(9)所示,式中H2A2代表P204萃取剂的二聚体。
由式(8)、式(9)可得出萃取反应的表观平衡常数K1ex、K2ex,见式(10)、式(11)。
结合式(2)分配比D,并将得到的式子两边取对数并重新排列后,有式(12)、式(13)。
利用斜率法[26]对相关数据进行计算并线性拟合,结果如图9所示。可以看出,lgD与lg[H2A2]呈线性关系且其斜率为2.29,R2=0.977[图9(a)];lgD与pH 呈线性关系且其斜率为1.27,R2=0.987[图9(b)]。对比式(12)与式(13)的相关系数发现,实验得到的斜率值均介于理论计算的两组系数值之间,这说明萃取过程中反应(8)与反应(9)均有发生,这与上述结果FeSO4+易被P204萃取相对应。
2.2.2 红外光谱分析
对萃取前和最佳萃取条件下的负载有机相进行FTIR 检测,结果如图10 所示。P204 分子的P—O—H、—P= = O特征吸收峰分别出现在1030cm-1与1685cm-1、1225cm-1处,萃取前曲线中1685cm-1处的峰不明显可能是由磺化煤油稀释造成的。其余位置如2960cm-1、2931cm-1、2861cm-1处出现的峰分别为—CH3、—CH2—。负载铁曲线中1685cm-1处P—O—H特征吸收峰消失,1030cm-1处的P—O—H特征吸收峰蓝移到1103cm-1处,说明P204萃取Fe3+时发生一次溶剂化反应,即分子式中的—OH 官能团直接与Fe3+发生了离子交换过程[25−26],生成的萃合物溶解于有机相中,H+则进入水相中,这也解释了萃取后水相pH降低的现象。另外,P204分子式的—P= = O特征吸收峰从1225cm-1处红移至1187cm-1,说明萃取过程中—P= = O 基与Fe3+发生了配位反应,因为P204 分子中磷酰基上边的氧含有孤对电子,Fe3+有空轨道,为两者的配位反应创造了条件,从而使Fe3+从水相转移至有机相中,生成的配位键使得原本的—P= = O 吸收峰位置发生改变,减小了吸收强度,造成曲线红移。因此P204萃取Fe3+时既存在离子交换过程,也存在配位反应过程。

图10 P204有机相及负载铁有机相的FTIR图
2.2.3 质谱分析
对萃取前和最佳萃取条件下的负载有机相进行ESI−MS检测,结果如图11所示。图11(a)中645.4599处的峰对应P204二聚体,这验证了P204主要以二聚体的形式存在。图11(b)中1345.88595、1987.71235处的峰分别为FeSO4A(HA)3、FeA3(HA)3,说明P204既能萃取溶液中游离Fe3+,也能萃取FeSO4+,这解释了图4的结论,也与化学计量学的分析验证相对应。其萃取过程的3D模型如图12所示,配合物FeA3(HA)3中Fe3+由六个键位组成,分别为三个P= = O 中的O与Fe3+形成的配位键和三个P—O—H 脱H 与Fe3+形成的共价键。FeSO4A(HA)3中Fe3+由两个共价键和三个配位键组成,一个共价键由P—O—H脱H与Fe3+形成,另一个共价键由SO42−与Fe3+形成,剩余三个配位键由P= = O中的O与Fe3+形成。

图11 ESI−MS分析
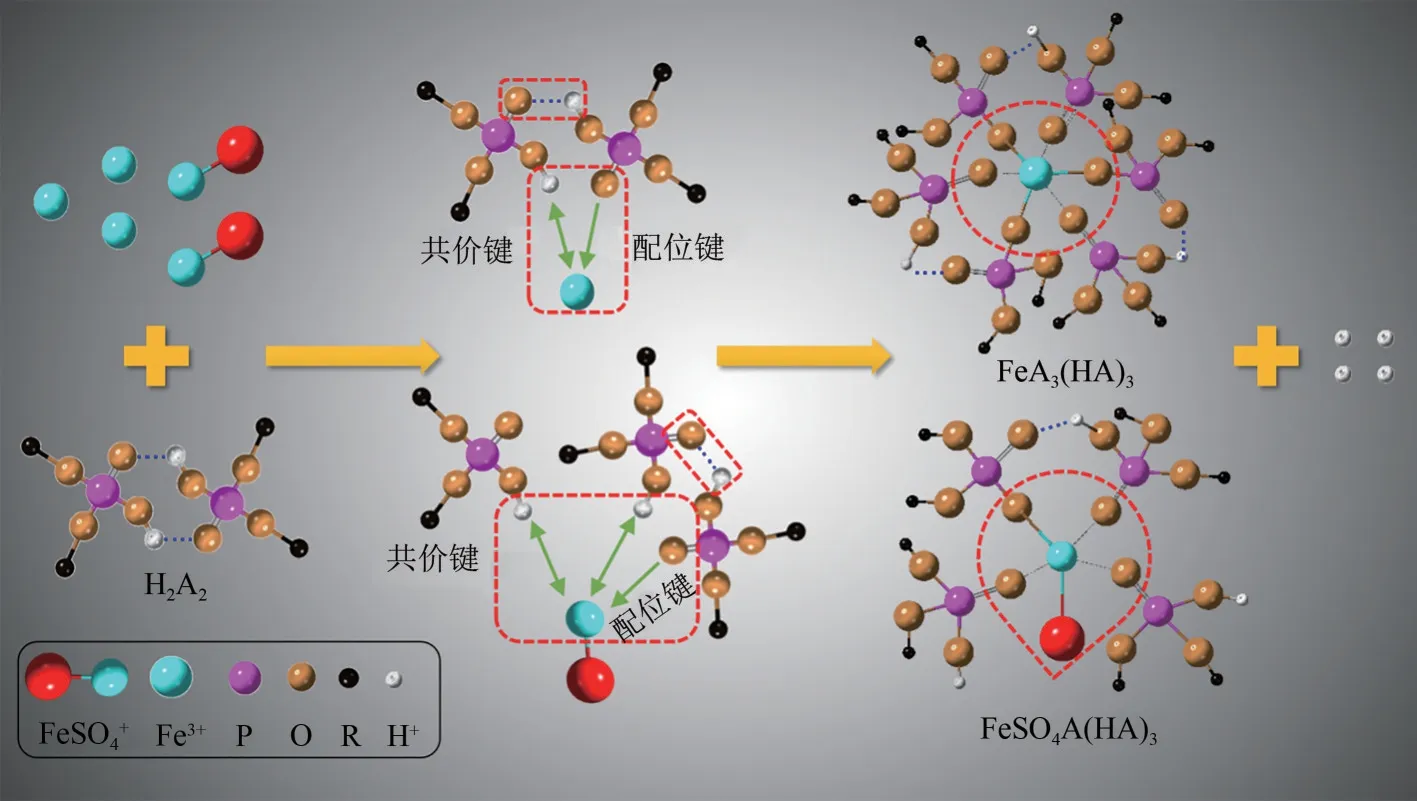
图12 萃取过程的3D模型
2.3 负载有机相反萃
在最佳萃取条件下,有机相中负载Fe3+浓度达3546.43mg/L,在相比O/A=1∶1、反萃温度35℃、时间10min、振荡频率190r/min 的条件下考察草酸浓度对负载铁、锌和铜反萃的影响,结果如图13所示。反萃液中未检测到Cu2+,一方面是因为负载有机相中Cu2+浓度仅0.07mg/L,另一方面草酸可能对Cu2+的反萃效果差。负载有机相中Fe3+和Zn2+的反萃率随草酸浓度的增加呈现出逐渐增大的趋势,草酸浓度为1mol/L时Fe3+的反萃率达到82.64%,此时Zn2+的反萃率为18.26%,负载有机相中剩余Zn2+浓度为2.872mg/L。当草酸浓度高于1mol/L 时,有机相中铁离子的反萃率上升趋势逐渐变缓,草酸浓度对反萃率的影响逐渐减弱。草酸是酸性较弱的有机酸,能与P204发生明显分层,草酸与Fe3+形成的络合物比P204与Fe3+的络合物更稳定,在草酸过量的情况下,使得有机相中的Fe3+经过反萃后以草酸铁络合物如[Fe(C2O4)3]3-、Fe(C2O4)+、Fe(C2O4)−2的形式进入水溶液中,反应方程如式(14)~式(16)所示。当[C2O2−4]/[Fe3+]>3 时,形成的络合物结构主要为[Fe(C2O4)]33-[27]。此外,在反萃液中加入10g/L 的BaCl2,有白色沉淀产生,继续加入0.5mol/L 盐酸后,白色沉淀不溶解,即反萃液中存在硫酸根,其可能发生式(17)的反应。以络合物[Fe(C2O4)3]3-、Fe(C2O4)+、Fe(C2O4)−2、[FeSO4(C2O4)]-形式存在的铁为后续光照条件下增值化利用——生产草酸亚铁奠定了基础[28−29]。此外,反萃过程中有0.627mg/L的Zn2+进入反萃液,可能是形成了含硫酸根的复杂化合物。值得注意的是,受溶解度的限制,在温度较低的情况下,草酸浓度高于1mol/L 时易结晶析出,导致反应的传质效率降低、设备孔径堵塞,因此草酸浓度不宜过高。取草酸浓度为1mol/L,既能保证较高的铁离子反萃率,也不会造成草酸的浪费,是比较适宜的草酸浓度。
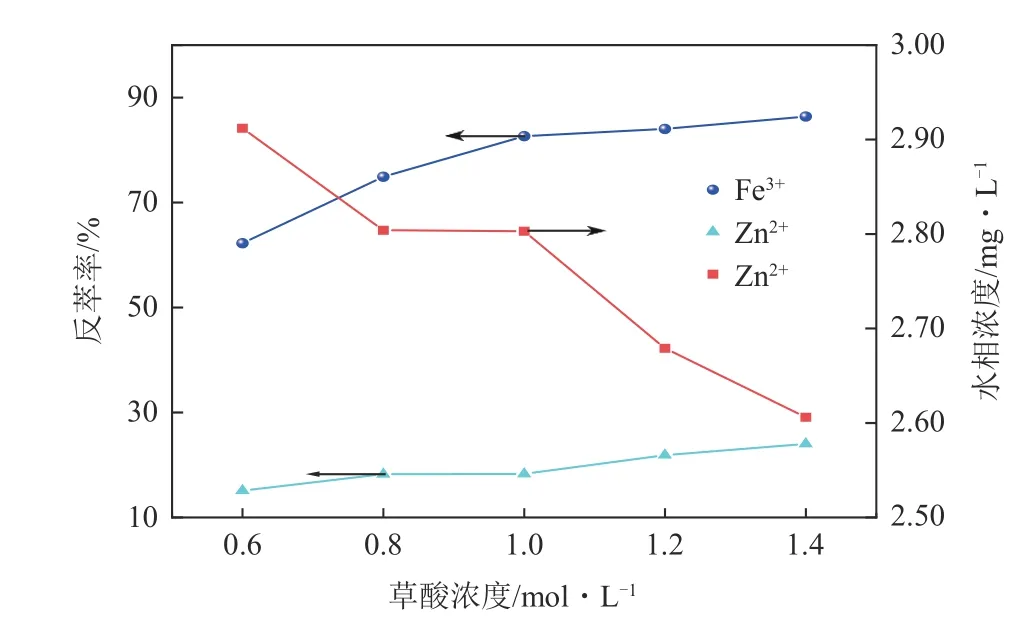
图13 草酸浓度对负载有机相反萃的影响
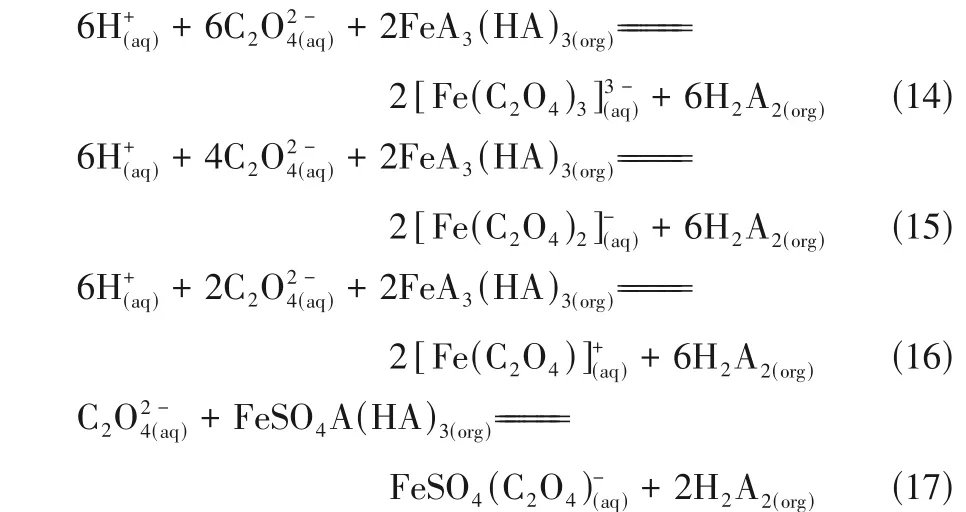
2.4 有机相的循环使用
将反萃后的P204−磺化煤油有机相在O/A=1∶1、振荡时间10min、振荡频率180r/min、温度25℃的条件下,重复5 次萃取−反萃氰化尾渣电解液,考察P204−磺化煤油的循环使用性,结果如图14 所示。可以看出,P204−磺化煤油有机相经过5 次循环后,电解液中Fe3+萃取率仍有96.45%,仅下降1.28%,仍具有良好的萃取效果,这为生成的P204−磺化煤油有机相循环使用创造了有利条件。草酸反萃后还剩余2.872mg/L的Zn2+存在于负载有机相中,由于负载有机相中Zn2+浓度低,考虑有机相循环多次后再用0.5mol/L盐酸进行洗脱除杂。另外,从反萃实验结果来看,反萃液中的Zn2+浓度也不是很高,在光催化制备草酸亚铁的过程中其并不会同时沉淀析出,从而影响草酸亚铁的纯度[30]。
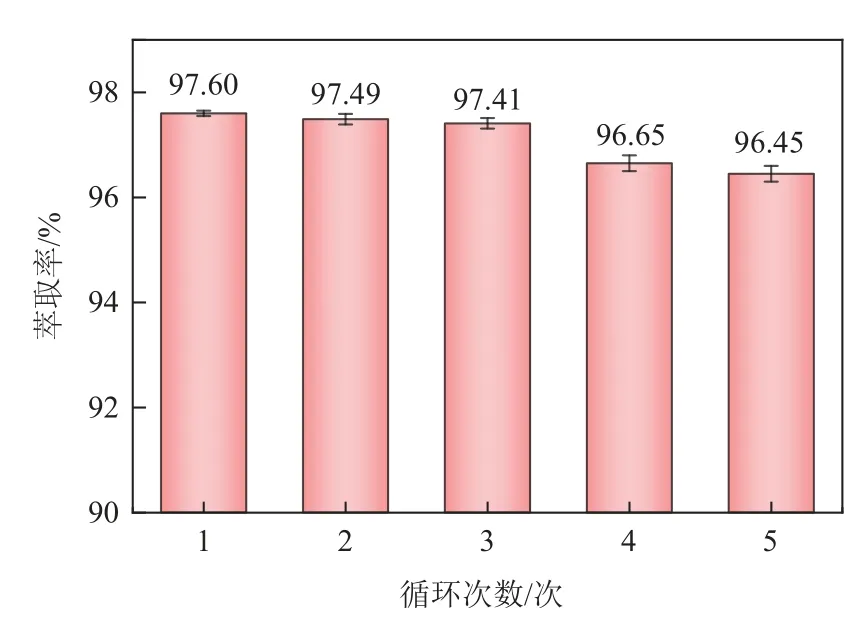
图14 有机相循环次数与Fe3+萃取率的关系
3 结论
用P204−磺化煤油作萃取剂能够有效萃取高硫酸氰化尾渣矿浆电解液中的Fe3+。在P204 体积分数为25%、O/A=1∶1、pH为1.5、时间10min、振荡频率为180r/min、萃取温度25℃的条件下,Fe3+萃取率可达到97.73%。P204 负载铁的饱和容量为21.57g/L,铁离子可富集5.86倍。P204萃取铁的反应方式为阳离子交换反应和配位反应,既可萃取游离Fe3+也能萃取FeSO4+,萃取生成配合物FeA3(HA)3、FeSO4A(HA)3。负载有机相采用草酸反萃,当草酸浓度为1mol/L 时铁反萃率可达到82.64%,反萃液中产物为[Fe(C2O4)3]3-、Fe(C2O4)+、Fe(C2O4)2-、FeSO4(C2O4)-,锌浓度仅0.628mg/L,未检测到铜,反萃后的有机相经5次循环使用后仍具有良好的萃取效果。本工艺采用P204−磺化煤油萃取体系、草酸反萃得到含铁络合物,为氰化尾渣无害化、资源化处理工艺的完善奠定了基础。
猜你喜欢
钢铁钒钛(2022年6期)2023-01-31
河南化工(2022年6期)2022-07-08
探索科学(学术版)(2021年3期)2021-07-12
理化检验-化学分册(2020年5期)2020-06-15
理化检验-化学分册(2020年5期)2020-06-15
石油地质与工程(2019年3期)2019-09-10
中国钼业(2019年2期)2019-01-19
世界有色金属(2018年17期)2018-11-20
西安建筑科技大学学报(自然科学版)(2014年2期)2014-11-12
无机盐工业(2014年3期)2014-03-20
