
Quinoline-based anti-MRSA agents: Current development,structure-activity relationships,and mechanisms
2023-02-18 01:54HongYaoLipingCuiHangLiuXueyuLiLinShenRuigeYangShangshangQinYongGuo
Chinese Chemical Letters 2023年12期
Hong Yao ,Liping Cui ,Hang Liu ,Xueyu Li ,Lin Shen ,Ruige Yang,∗ ,Shangshang Qin,∗,Yong Guo,c,∗
a College of Veterinary Medicine,Henan Agricultural University,Zhengzhou 450046,China
b School of Pharmaceutical Sciences,Zhengzhou University,Zhengzhou 450001,China
c Hunan Province Cooperative Innovation Center for Molecular Target New Drug Study,School of Pharmaceutical Science,Hengyang Medical School,University of South China,Hengyang 421001,China
Keywords:Quinoline analogue Methicillin-resistant Staphylococcusaureus Antibacterial activity Structure–activity relationship Antibacterial mechanism
ABSTRACT Methicillin-resistant Staphylococcus aureus (MRSA),the most common pathogen in hospital and community environments,can cause serious and even fatal infections.The antibiotics currently used for clinical treatment of MRSA have developed resistance,and there is an urgent need to develop new antimicrobials to treat infections caused by MRSA strains.Quinoline analogues play an important role in the development of antimicrobials.Herein,we discussed the current development of antibacterial activities of quinoline analogues,mainly for anti-MRSA activity,and their structure–activity relationships (SARs) from the perspective of using the quinoline nucleus to search for novel potential anti-MRSA candidates.Additionally,the mechanisms of some representative quinoline analogues against MRSA were clarified.Altogether,this review could provide further insights for the rational development of quinoline-based antibacterial drugs,especially against MRSA.
1.Introduction
Antibiotics are primarily used to treat a variety of bacterial infections,but due to overuse and misuse of antibiotics,bacteria have become resistant to currently approved marketed drugs[1,2].The spread of antimicrobial resistance (AMR) has made once treatable diseases deadly again,and bacterial infections have become a widespread global health risk [3–6].Staphylococcus aureus(S.aureus) is a round,Gram-positive bacteria that causes a variety of diseases ranging from skin and soft tissue infections to severe pneumonia,sepsis,and meningitis.Currently,more than 60% ofS.aureusisolates are methicillin-resistantS.aureus(MRSA)[7–12].Methicillin resistance inS.aureusstrains is caused by theinvivogene encoding PBP2a,which has an ultra-lowβ-lactam affinity and is capable of cell wall synthesis at normally lethalβ-lactam concentrations to complete the bacterial reproductive growth,hence MRSA is also known as a superbug [13,14].And as the last line of defense,vancomycin,MRSA has also developed resistance to it.Therefore,there is an urgent need to develop new antimicrobials with anti-MRSA potency [15,16].
Among the heterocyclic derivatives containing nitrogen atoms,quinolines are used in drug design and discovery due to their wide range of biological activities [17,18].Quinolines have been reported to have anticancer [19],antimalarial [20],antihypertensive [21],anti-inflammatory and analgesic [22,23],antibacterial[24],anti-human immunodeficiency virus (HIV) [25],antitubercular [26],anti-cardiovascular activities [27],and inhibition of tyrosine kinase [28] properties.At present,the drugs such as ofloxacin,norfloxacin,ciprofloxacin,and sparfloxacin,which have been approved for marketing to treat various bacterial infections,all contain a quinoline backbone in their structures [29,30].Although there are some reviews on the biological activity of quinolines,a review on the antibacterial activity of quinolines against MRSA has not been reported.Herein,we reviewed the structure–activity relationships (SARs) and different mechanisms of potential antibacterial effects of quinoline analogues (Fig.1) against MRSA strains in recent two decades to provide insights into the development of new potent quinoline-based antimicrobial agents.
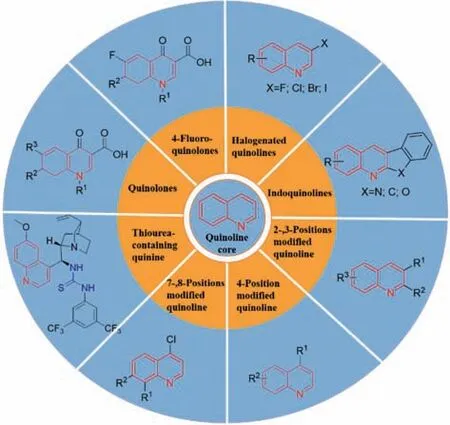
Fig.1. Structures of quinoline analogues related to anti-MRSA activities.
2.Quinolones
4-Quinolones such as nemonoxacin [31,32] and delafloxacin[33] play a key role in the clinical anti-MRSA infection treatment.However,MRSA has developed resistance to 4-quinolones [34],so it is becoming increasingly urgent to improve the efficacy of 4-quinolones.Modifications of the C-7 position of 4-quinolones are thought to be beneficial for improving antibacterial potency,solubility,and safety [35,36].
HT-61 (1,Fig.2),is a quinolone derivative with a molecular weight of about 400 Daltons that is particularly active againstS.aureus,including MRSA [37,38].Compound1not only acted on reproductive bacteria with minimum inhibitory concentrations(MICs) higher than those of marketed antibiotics but also displayed significant efficacy against non-reproductive bacteria.The MICs of1against 45 MRSA isolates were 4–8 μg/mL and its minimum stationary phase-cidal concentration 50 (MSC50) values against 103 MRSA isolates were 1.5–7.5 μg/mL.Compound1was significantly more potent than daptomycin,which required 50 μg/mL to completely kill fixed-stage MRSA.Additionally,compound1can target bacterial cell membranes and act on the plasma membrane of bacterial cells by disrupting the membrane potential ofS.aureus,resulting in the release of cellular contents.Moreover,compound1in combination with several antibiotics against clinical MRSA strains was also investigated,and the results showed that compound1had significant synergistic activity when it combined with gentamicin,neomycin,or chlorhexidine.
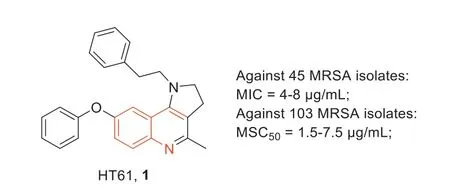
Fig.2. Structure of compound 1 and its anti-MRSA activity.
A few tricyclic fluoroquinolones,[1,2,4]triazolo[3,4-h][1,8]naphthyridine-8-one-7-carboxylic acid derivatives2a–h(Fig.3),whose C-8 positions contained a functional Mannich base part,were evaluated for theirinvitroantibacterial activities against MRSA [39].The results showed that the Mannich base derived from the cycloaliphatic amine donors (2d–h) exhibited strong antibacterial activity against MRSA,with MIC values of 0.5–2 μg/mL,and its potency was 2–8 times stronger than that of ciprofloxacin.Among them,especially compound2ghad the best antibacterial activity with an MIC value of 0.5 μg/mL,which was 8 times higher than that of ciprofloxacin (Fig.3).
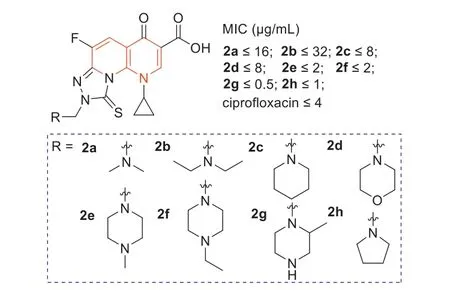
Fig.3. Structures of compounds 2a–h and their anti-MRSA activities.
The new benzimidazole quinolones drugs displayed promising antibacterial action [40].Antibacterial evaluationinvitroshowed that most of the title benzimidazole quinolones exhibited good antibacterial activity against the tested strains,especially against MRSA,even better than the reference drugs [40].For example,methylene-bridge linkers,halogenated benzyl derivatives,NH2CH2linkers and benzimidazole complexes directly linked to quinolone nucleus displayed good anti-MRSA activity,with MIC values of 0.125–0.5 μg/mL.They were more active than chloramphenicol (MIC=16 μg/mL),norfloxacin (MIC=8 μg/mL),ciprofloxacin(MIC=2 μg/mL),and clinafloxacin (MIC=1 μg/mL).The anti-MRSA activity of methylene-linked compounds3a,3b,3c,and3d(Fig.4,MIC=0.125 μg/mL) was 8–64 times higher than that of norfloxacin,ciprofloxacin,and clinafloxacin.Compound3dshowed rapid bactericidal activity,and the number of viable bacteria decreased by more than 3 logs (CFU/mL) within an hour at a concentration of 6×MIC.Moreover,3dnot only can inhibit the formation of biofilm but also can disperse the formed bacterial biofilm,while exhibiting low toxicity to mammalian cells and not inducing bacterial resistance.The results of SARs indicated that when the benzimidazole and quinolone nucleus were linked by a methylene linkage,there was a significant effect on the enhancement of antibacterial activity (Fig.4).
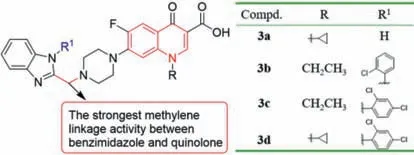
Fig.4. Structures of compounds 3a–d.
In 2015,some novel quinolone derivatives based on levofloxacin as the core were synthesized by Huangetal.[41].The antibacterial activity of4a–d(Fig.5,MIC=1 μg/mL) was 4 times greater than that of moxifloxacin (MIC=4 μg/mL),32 times stronger than that of levofloxacin (MIC=32 μg/mL) and as isoenergetic as vancomycin(MIC=1 μg/mL).Compound4bhad better solubility (5.33 mg/mL)and lower toxicity than levofloxacin.In addition,the lethal dose 50(LD50) of4bwas 1402 mg/kg,while the LD50of levofloxacin was 358 mg/kg.
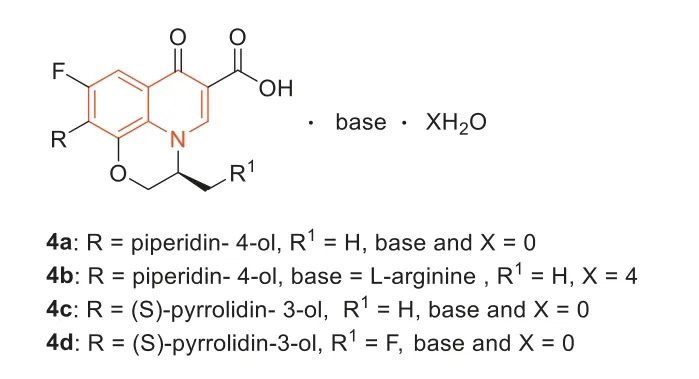
Fig.5. Structures of compounds 4a–d.
Zhangetal.[42] reported the development of fluoroquinolone derivatives containing 3-alkoxyimino-4-(cyclopropylanimo)methylpyrrolidine moiety and evaluated their biological activities.Among these fluoroquinolone derivatives,compound5a(Fig.6) exhibited the strongest antibacterial activity against MRSA with an MIC value of 2 μg/mL,4–64 times higher than ciprofloxacin and levofloxacin (MIC: 8–128 μg/mL) against MRSA.The SARs indicated that the introduction of methyl at 3-position of the pyrrolidine ring increased antibacterial activity against Gram-positive bacteria,which was consistent with the results reported by Yunetal.[43].The compounds from the two research groups had the same parent nuclear structure,and compound5bsynthesized by Yunetal.[43] displayed goodinvivoefficacy against MRSA in a mouse infection model (Fig.6).
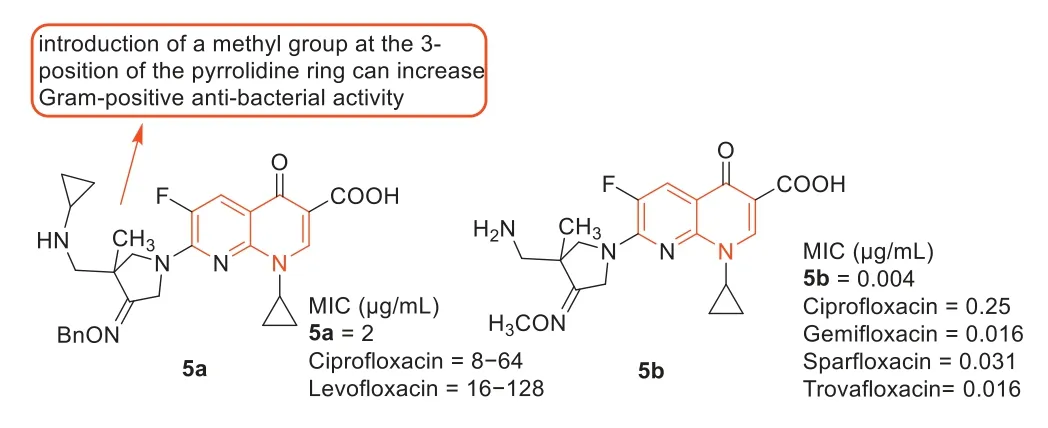
Fig.6. Structures of compounds 5a, 5b,and their anti-MRSA activities.
Cuietal.[44] inserted the triazole ethanol moiety into the N-1 position of quinolones to change the different substituents on the benzene ring of quinolones,and prepared a new class of quinolones triazoles.Most of these new quinolone triazoles can effectively inhibit MRSA growth (MIC=0.5-16 μg/mL),which were more effective than clinical drug chloramphenicol(MIC=16 μg/mL).Compounds6a,6b,and6c(Fig.7) with MIC values of 1 μg/mL were 8 and 16 times more active than norfloxacin (MIC=8 μg/mL) and chloramphenicol (MIC=16 μg/mL),respectively.In particular,7-trifluoromethyl intermediate6d(Fig.7,MIC=0.5 μg/mL),as a new potential anti-MRSA candidate,exhibited 16 and 32 times higher antibacterial activity than the standard drugs norfloxacin and chloramphenicol,respectively.In addition,Cui’s [45] group also synthesized a series of novel 3-aminothiazole quinolones analogues as antibacterial agents.Among these 3-aminothiazol-quinolones,3-(2-aminothiazol-4-yl)-7-chloro-6-(pyrrolidine-1-yl) quinolone6e(Fig.7) showed significant antibacterial activity against MRSA at low concentrations(MIC=0.8 μg/mL).Its antibacterial activity was 13 and 27 times higher than that of norfloxacin and chloramphenicol,respectively.Moreover,6ehad low cytotoxicity to liver cells,strong inhibition of DNA cyclotron enzyme,and a wide antimicrobial spectrum,including MDR strains.Notably,the active molecule6ealso induced bacterial resistance more slowly than norfloxacin.Analysis of the SARs (Fig.7) revealed that the 2-aminothiazole fragment at C-3 position of quinolones increased antibacterial potency and could replace the carboxyl group of quinolones.The introduction of some electron-donating substituents,such as methyl,methoxy,or alicyclic amino groups,at 6-and 7-positions of quinolones was particularly beneficial for anti-Gram-positive bacteria.The antibacterial evaluation showed that 7-chloroquinolones had better inhibitory activity against MRSA than 7-fluoroquinolones.Additionally,the substituents on the benzene ring of quinolones also affected their antibacterial potency.
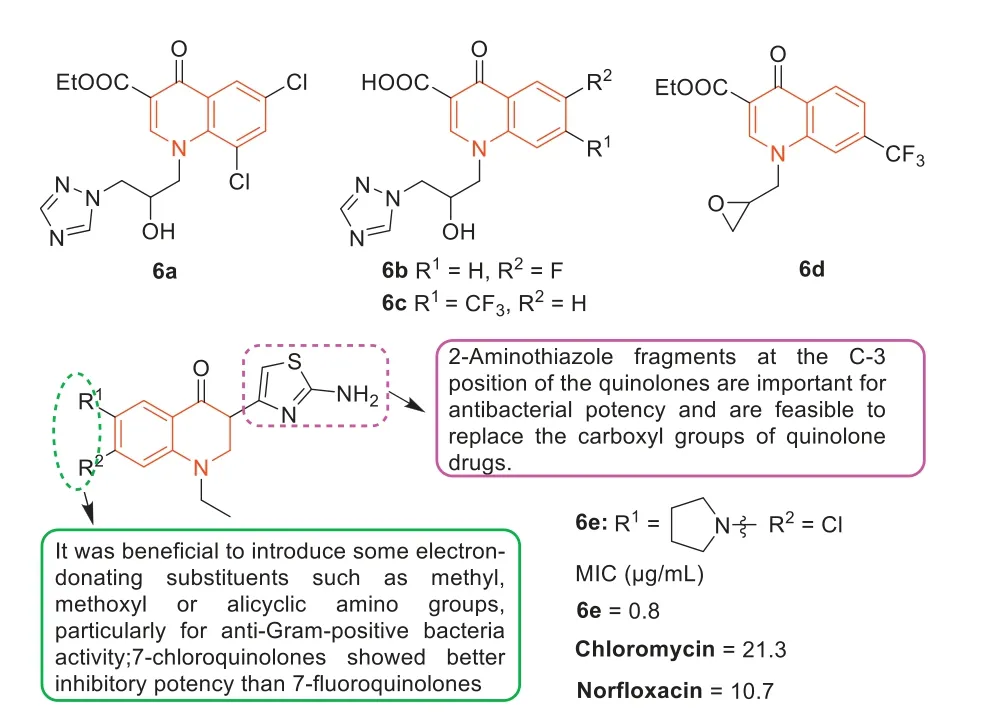
Fig.7. Structures of compounds 6a–e and their SARs.
In 2020,Maryametal.synthesized some new fluoroquinolones with ciprofloxacin and sarafloxacin as the core skeletons [46].All the compounds synthesized from ciprofloxacin derivatives showed significant anti-MRSA activity,with MIC values ranging from 0.016 μg/mL to 0.29 μg/mL.The antibacterial activity of compound7aagainst MRSA (MIC=0.016 μg/mL) was 60 times higher than that of the positive control ciprofloxacin (MIC=0.49 μg/mL)(Table 1).This meant that the presence of methoxy at the R1position produced beneficial anti-MRSA activity.All the synthetic sarafloxacin derivatives exhibited approximately the same activity as the parent sarafloxacin against Gram-positive bacteria.Especially compound7bwith a methoxy group at the R1position showed the most promising antibacterial activity against MRSA(MIC=0.125 μg/mL),which was twice as high as the positive control sarafloxacin (MIC=0.25 μg/mL) (Table 1).
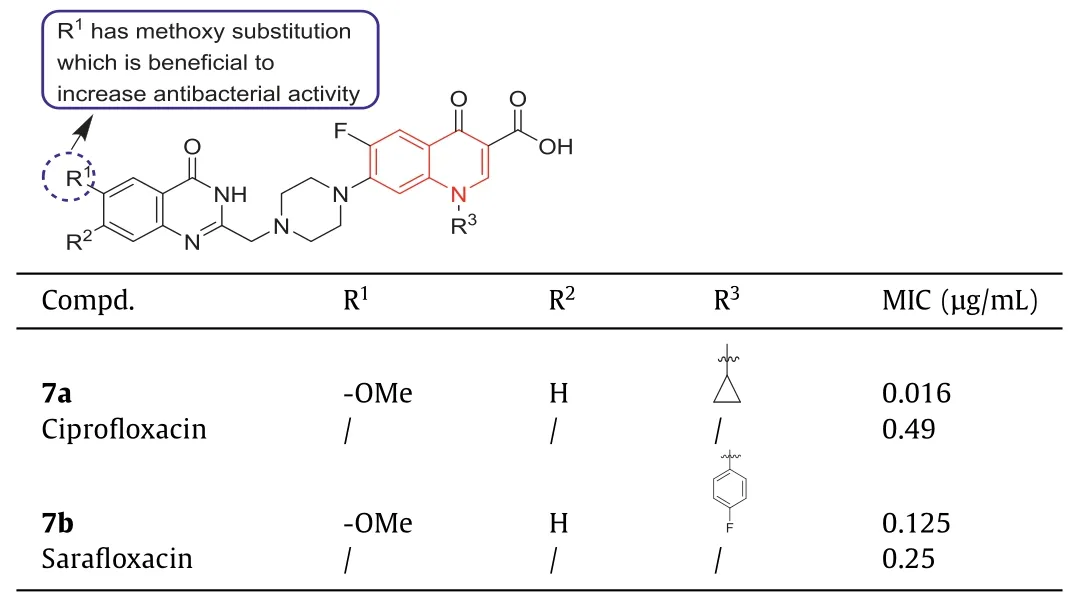
Table 1 Structures of compounds 7a, 7b,and their anti-MRSA activities.
In 1987,isothiazoloquinolones (ITQs),originally reported by Chu and colleagues [47,48],were more potent than quinolones againstS.aureus.The best represented of ITQs was the ciprofloxacin analogueA-628249(Fig.8).Wiles and Wangetal.[49–52] synthesized ITQ-related compounds containing functionalized aryl and heteroaryl groups connected by a C–C bond at 7-position and structurally modified at 6-,8-,and 9-positions,such as compounds8a,8b,and9(Table 2).These compounds exhibited excellentinvitroantibacterial activity against MRSA as well as strong inhibitory activity against DNA rotamases and topoisomerase IV.In 2011,a series of 8-methoxy ITQs with a 7-aminocyclic ring was synthesized by Kimetal.[53].The structures of10a(R,S) and10b(R) are shown in Fig.9.The MICs of10aand10bagainst MRSA and their inhibitory activities against target enzymes fromS.aureus,wild-type topoisomerase IV and DNA rotamase,and the corresponding enzymes fromStaphylococcalmutants expressing fluoroquinolone resistance are outlined in Table 3.Compounds10a(R,S) and11b(R) exhibited potent anti-MRSA activity with an MIC value of 0.06 μg/mL.Notably,compared to ciprofloxacin,10b(R)increased inhibition of wild-type DNA gyrase activity by 91-fold,while10a(R,S) increased inhibition of wild-type topoisomerase IV activity by 75-fold.Similar improvements were observed for10b(R) and10a(R,S) inhibition of mutant DNA gyrase,with>48-fold and>62-fold increases,respectively.Compounds10b(R) and10a(R,S) increased the inhibition of mutant topoisomerase IV by 95-fold and 121-fold,respectively.The above data suggested that 8-position with methoxy could increase anti-MRSA activity and reduce cytotoxicity.
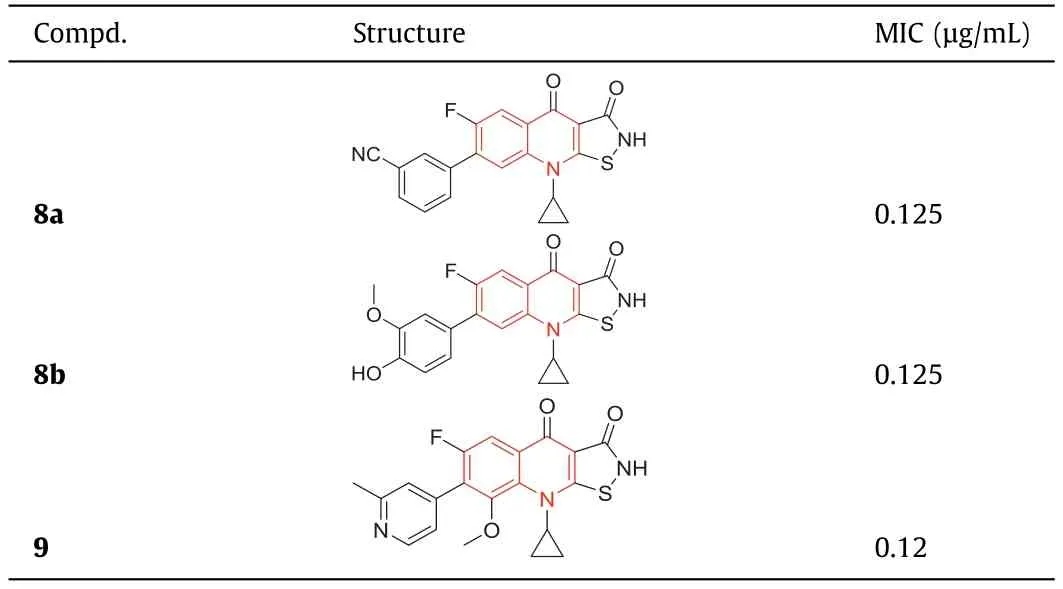
Table 2 Structures of compounds 8a, 8b, 9 and their anti-MRSA activities.
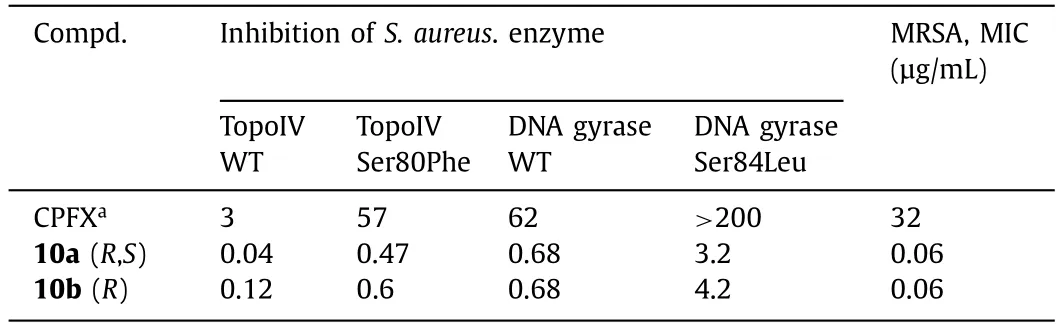
Table 3 In vitro activities of compounds 10a (R,S) and 10b (R).
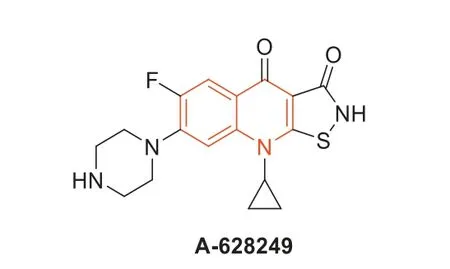
Fig.8. Structure of compound A-628249.
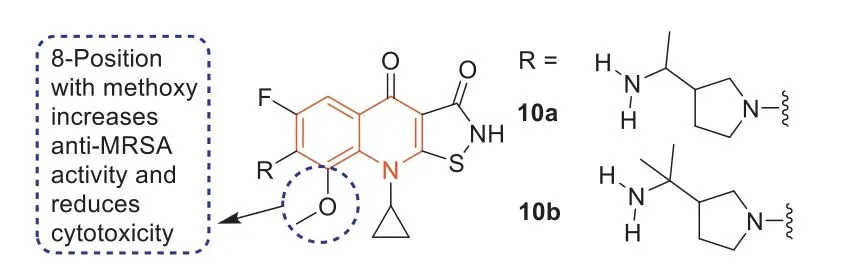
Fig.9. Structures of compounds 10a and 10b.
3.Halogenated quinolines
In 2010,O’Donnelletal.[54] performed an antimicrobial evaluation of haloquinoline derivatives,in which 4-hydroxy-3-iodoquinolin-2-one (11) showed strong to excellent antibacterial activity against eight clinical MRSA isolates (Table S1 in Supporting information).For clinical isolates EMRSA-16,CWMRSA,and HMRSA,the MIC value of11was equal to or lower than that of vancomycin,an antibiotic used in the clinical treatment of MRSA infection.While the antibacterial activity of11against other clinical MRSA isolates was better than vancomycin (MIC=0.78–1.56 μg/mL).
In 2014,Abouelhassan’s group [55] synthesized a library of 21 quinoline analogues and evaluated their antimicrobial activities(Fig.10).Among these 21 quinoline analogues,some quinoline analogues showed effective activity against MRSA-2.The quinoline derivative12a–e(Fig.10) exhibited very effective biofilm dispersion against MRSA-2 (EC50=2.80 μmol/L).In particular,quinoline derivative12c(EC50=2.06 μmol/L) exhibited the most effective biofilm dispersion activity.The 2-position of the halogenated quinoline (HQ) scaffold played an important role in the corresponding antibacterial activity of the HQ analogues.Compared with quinoline derivative12a,bromoxyquinoline12f(Fig.10) had a hydrogen atom at 2-position,and12ahad a methyl group at 2-position of the HQ scaffold (Fig.10).This monomethyl difference resulted in a 16-fold increase in the antibacterial activity of12aagainstStaphylococcalpathogens and a 128-fold elimination of antibacterial activity against the Gram-negative pathogensAcinetobacterbaumannii.In addition,12acan also eradicate MRSA biofilms.Based on the potent anti-MRSA activity,they subsequently synthesized a series of HQ analogues,which had different alkylation groups at the 2-position of the HQ scaffold [56].In addition to 2-alkylated HQ analogues,they also designed an alternative pathway to synthesize reductive amination HQ analogues by reducing amination at 2-position of the HQ scaffold with various amines and anilines [56].Some HQ analogues showed effective antibacterial activity,such as13aand13b(Fig.10).It is worth mentioning that the MIC values of14a(Fig.10) and12a(MIC≤0.78 μmol/L) against MRSA-2 were identical.Among these HQ analogues,14b(Fig.10) exhibited the highest anti-MRSA activity with MIC of 0.39 μg/mL,and it did not show hemolysis toward red blood cells (RBC) at 200 μmol/L.In general,HQ analogues containing aniline halide groups displayed good to highly potent antibacterial activity,while HQ analogues containing alkyl and methoxylaniline groups usually showed low antibacterial activity.For example,13b[minimum biofilm eliminating concentration(MBEC)=93.8 μmol/L] and14b(MBEC=93.8 μmol/L) displayed improved biofilm eradication activity against MRSA-2 compared with12a(MBEC=188 μmol/L).These synthesized HQ analogues were 20 times more potent as MRSA-2 biofilm eradicators than current anti-MRSA therapeutic agents including vancomycin,daptomycin,and linezolid.Moreover,they displayed low hemolysis and low cytotoxicity to HeLa cells [56].Eighty percent of bacterial infections are related to biofilm,and halogenated 8-hydroxyquinolines had the characteristics of biofilm degradation.Accordingly,they used Friedlander reactions to synthesize a series of halogenated quinolines,capable of eradicating bacterial biofilms while exhibiting minimal mammalian cytotoxicity and hemolytic activity [56].In preliminary MIC assays,some HQ compounds exhibited higher antimicrobial activity compared to12a.HQ compounds15aand15b(Fig.10) showed improved activity against MRSA-2,with MIC of 0.39 and 0.59 μmol/L,respectively,and15bproved to be one of the most effective biofilm eradicators ever reported against MRSA (MBEC=3.9–23.5 μmol/L) [57–59].Antimicrobial enhancement is defined as the ability of a non-growth inhibitory enhancer [the plant-derived chemical gallic acid (GA)] to reduce the MIC value of HQ by more than 4-fold.It is proven that combination therapy can improve antibacterial activity.Abouelhassanetal.[60] also evaluated enhancement assays in combination with GA against four clinical MRSA (MRSA-ATCC,MRSA-1,MRSA-2,and SA-156).Compounds12f–h(Fig.10) had a 2–1000-fold enhancement effect when combined with GA.When12fwas combined with GA,its activity was increased by 1000-fold potentiation against MRSA SA-156.They also studied the combination of GA with conventional antibacterial agents,such as ciprofloxacin and vancomycin.However,none of these antibacterial agents were enhanced by GA.These results indicated that the unique antibacterial mechanism of the HQ compounds was different from that of traditional antibiotics.HQ compounds operated through a metal(II) dependent mechanism and unlike conventional antibiotics,exhibited good cytotoxicity when tested in HeLa cells in lactate dehydrogenase (LDH) release assay.In summary,Abouelhassanetal.[60] found that the selective combination of GA and HQ small molecules had strong antibacterial activity against a variety of pathogens,including multidrug-resistant clinical isolates.The combination therapy has also been effective in eradicating MRSA biofilms.The phytochemical-HQ combination provided a promising new platform for the development of clinically useful antibacterial combination therapy [60].
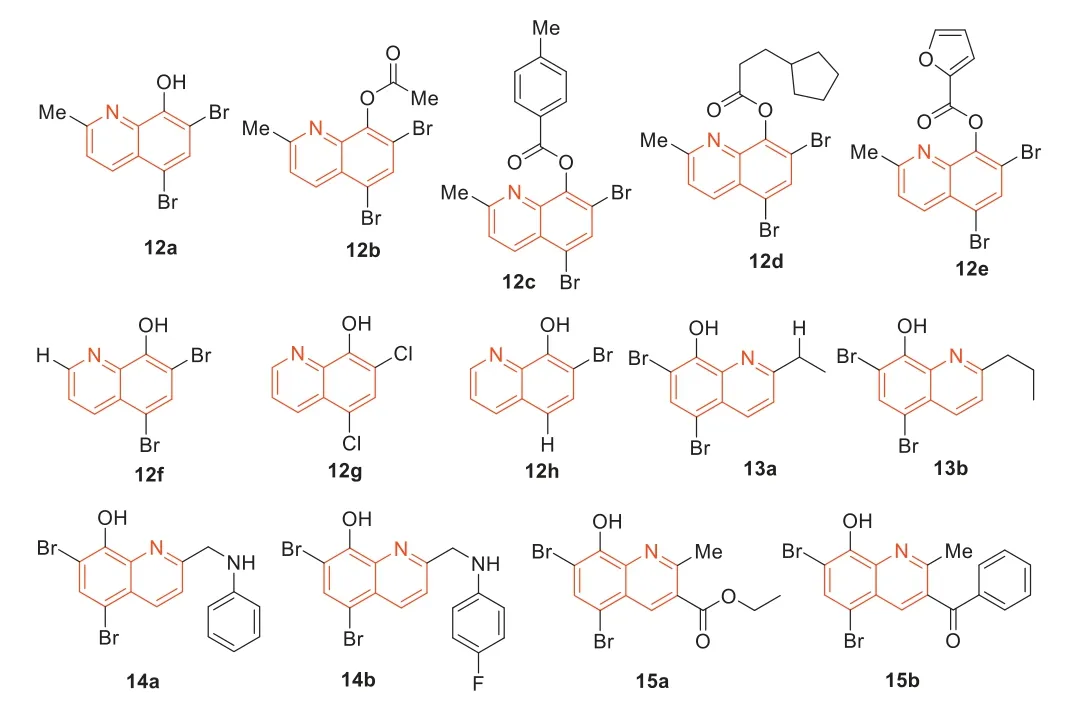
Fig.10. Structures of the halogenated quinolines 12–15.
4.Indoquinolines
The indole part is an important basic subunit for a large number of natural products and drugs [61],and indole-containing molecules have been reported to possess some antibacterial activity [62,63].Moreover,indoquinoline analogues also play an important role in antimicrobial activity.
Under 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ)-mediated oxidation conditions,Challaetal.[64] prepared a library of indolo[2,3-b]quinolines,chromeno[2,3-b]indoles,and 3-enyloctadiols using readily available 3,3'-diindolylmethane (DIM)(the synthetic route of the representative compounds16aand16bwas shown in Scheme S1 in Supporting information).The MIC and minimum bactericidal concentration (MBC) of indoloquinolines against four different methicillin-resistantS.aureusstrains (three of which were clinical isolates) were determined.The results showed that16aand16bpossessed excellent activity against methicillin-sensitiveS.aureusand MRSA (Table 4).Methicillin was active against susceptible strains,and the MIC value was 1 μg/mL,but it had MIC values of 32 μg/mL to>64 μg/mL for all MRSA strains.This result indicated that the MRSA strain was highly resistant to methicillin.In contrast,compounds16aand16bshowed similar activity against both drug-sensitive and drug-resistant strains,while the MIC values of these two compounds against methicillin-sensitive strains were 1 μg/mL.For MRSA strains,the MIC values of16aand16branged from 1 μg/mL to 4 μg/mL,and the MBC values of16aand16branged from 2 μg/mL to 8 μg/mL,while methicillin had an MIC value of>64 μg/mL in all tested MRSA strains (Table 4).The results manifested that compounds16aand16bwere selective anti-MRSA agents,and their activitiesagainst MRSA were better than that against other bacteria.At the concentration of 16 μg/mL,both compounds16aand16bcompletely killed MRSA within 360 min.
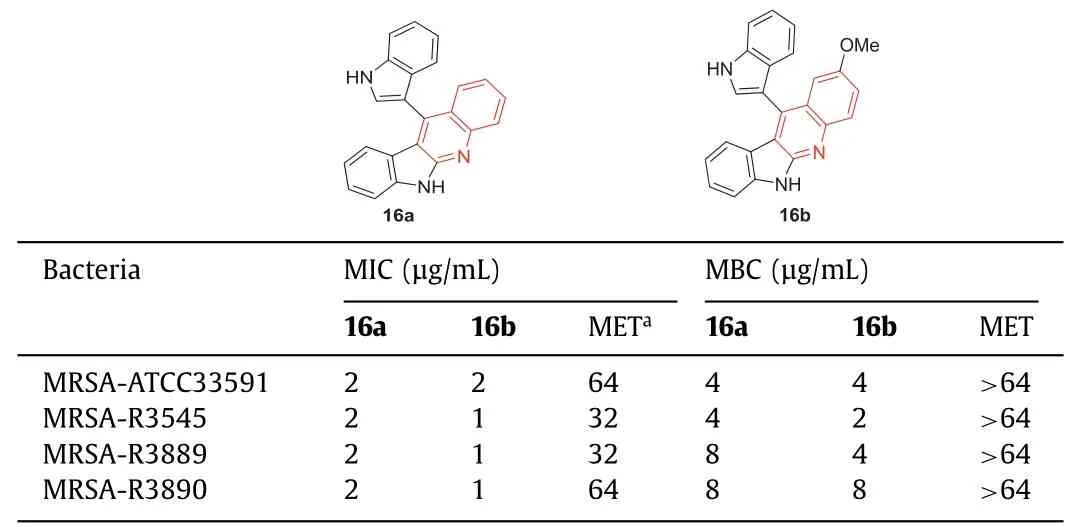
Table 4 Structures of compounds 16a, 16b,and their anti-MRSA activities.
In 2017,the antibacterial activity and mode of action of someN-methylbenzofurano[3,2-b]quinolones andN-methylbenzoindolo[3,2-b]quinoline derivatives containing quaternary pyridine centers were studied by Sunetal.[65].The results showed that all compounds had good antibacterial activity compared to the 11 clinical antibiotics evaluated in this study.In general,the antibacterial activity of 11-aniline-substituted 5-N-methyl-10-Hindole[3,2-b] quinolinium derivatives (b series) was slightly higher than that of 11-aniline-substituted 5-n-methylbenzofuran[3,2-b]quinoline derivatives (a series) (Fig.11).This result may be due to the interaction between the imine in the quaternary pyridine center and the amino acids in the filamentous temperaturesensitive protein Z (FtsZ).In addition,the addition of lipophilic groups at the 4-position of 11-aniline slightly enhanced the antibacterial activity of theseN-methyl quaternary ammonium derivatives.Among these derivatives,17b1,17b2,and17b3(Fig.11)were the most effective against MRSA,with MIC values of 2 μg/mL,and their antibacterial activities were about 100 times higher than that of berberine and methicillin.In the guanosine triphosphatase(GTPase) assay,17b2inhibited the GTPase activity of FtsZ in a dosedependent manner.The binding of the quinoline derivatives to the C-terminal domain gap interfered with the GTPase activity of FtsZ,and then disrupted the formation of FtsZ polymer,leading to abnormal cell division and cell death.
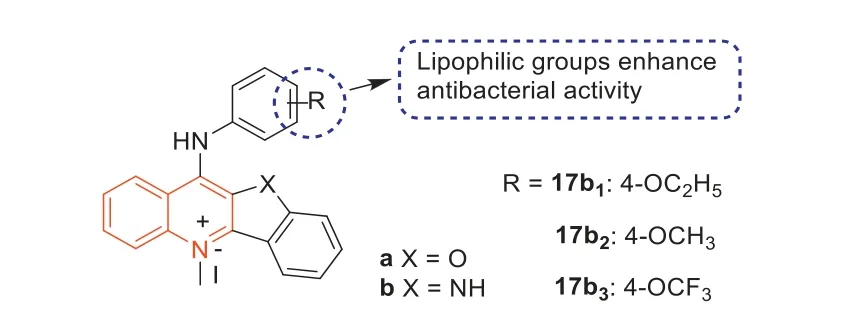
Fig.11. Structures of compounds a, b, 17b1, 17b2,and 17b3.
The ester analogues in 9-bromo-substituted indolylquinoline-5,12-dione derivatives showed strong antibacterial activity against Gram-positive strains,especially against MRSA [66].The MIC values of compounds18a,18b,and18cagainst MRSA were as high as 0.063 μg/mL,which was 16 times higher than that of vancomycin.Compound18dexhibited the best activity with an MIC value of 0.031 μg/mL,which was 32 times higher than vancomycin(Table 5).Compound18ahad a strong inhibitory effect on the superhelix activity of DNA rotation ofE.coliand the relaxation activity of topoisomerase IV ofS.aureus,exhibiting a mechanism similar to ciprofloxacin [67,68].Although compound18adisplayed good anti-MRSA activityinvitro,its bioavailability in mouse models was very low,probably because of its low water solubility.SARs (Fig.12) studies have revealed that: (1) The substitution of 9-position with bromine can enhance the antibacterial activity,but the substitution of 9-position with chlorine shows a lower activity [69].(2) Although 7-fluorinated analogues exhibited strong antibacterial activity with the same MIC value as compound18a,it had high cytotoxicity,indicating that it may not be a good choice for further development of good antibacterial agents [70].(3) The substitution of alkyl,amino,and carbonyl groups at the 6-position resulted in a significant decrease in antibacterial activity,which may be attributed to their reduced solubility.(4) The presence and position of nitrogen atoms in the ring were important for activity.The antibacterial activity of analogues with nitrogen atoms at 4-position or without nitrogen atoms decreased significantly.On the other hand,substitution of the 6-position by the alkoxycarbonyl group was important for enhancing the antibacterial activity.The modification of compound18awas guided by SARs and 28 indolylquinoline-5,12-diketone derivatives were synthesized to obtain the derivatives with higher anti-MRSA activity and good water solubility.Compound18a1exhibited strong activity against clinical MRSA strains,with MIC50and MIC90values lower than 0.0078 μg/mL.Compound18a2was soluble in water with a solubility of 1.98 mg/mL and also displayed strong activity against clinical MRSA strains with an MIC50value of 0.063 μg/mL and an MIC90value of 0.125 μg/mL,which was 16 times higher than vancomycin(Table S2,Supporting information),suggesting that it could be further developed as an anti-MRSA lead compound.
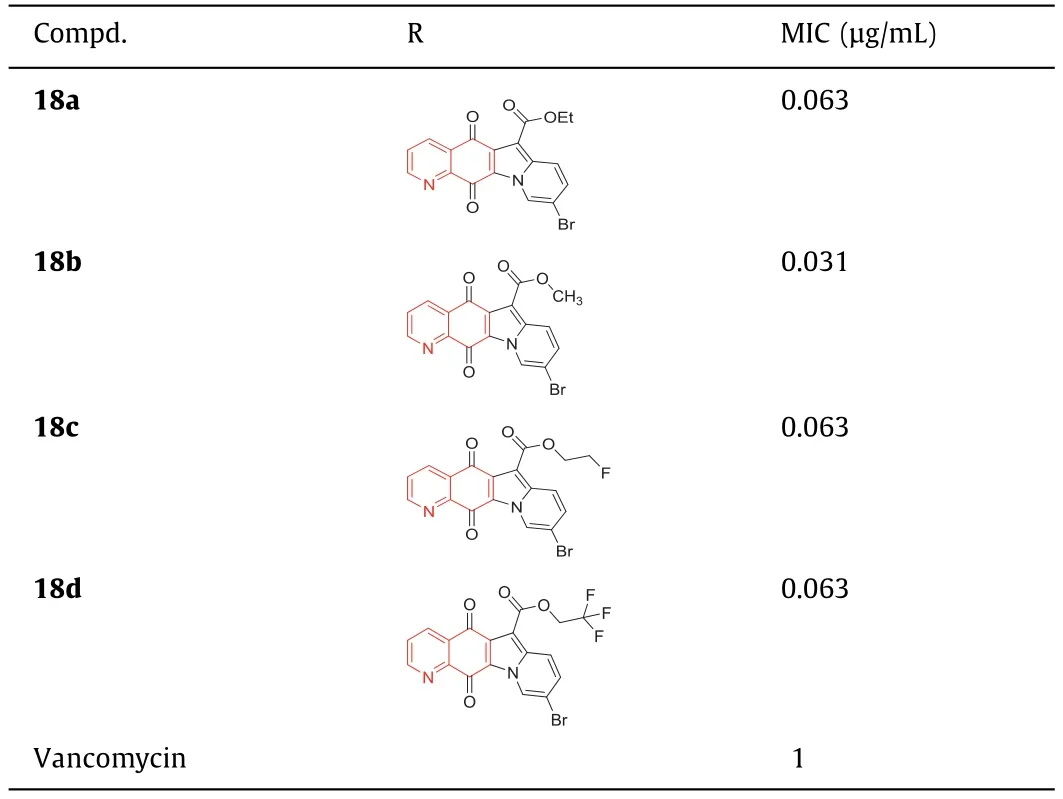
Table 5 Structure of compounds 18a–d and their anti-MRSA activities.
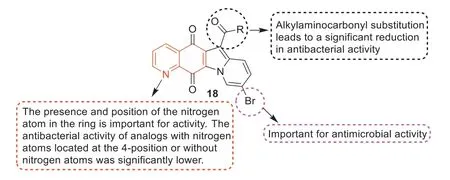
Fig.12. Summary of SARs study of compound 18.
In 1990,compound19was first synthesized by Yamatoetal.[71] for the study of anti-tumor activity (Table 6).Zhaoetal.[72–75] believed that compound19,containing the new skeleton of indoquinoline and its analogues,might have antibacterial activity and then measured the anti-MRSA activity of compound19against MRSA strains OM481 and OM584.The results showed that compound19had no activity against MRSA.Subsequently,they determined the anti-MRSA activity of analogues of compound19and found that compound20had an anti-MRSA activity with MIC of 8 μg/mL (against MRSA OM481) and 16 μg/mL (against MRSA OM584).Afterwards,Zhaoetal.[76] synthesized indolo[3,2-b]quinoline analogues,4-(acridine-9-amino)phenol hydrochloride(21),benzofurano[3,2-b]quinoline,and indolo[1,2-b]quinoline based on the indolo[3,2-b]quinoline backbone of lead compound20and investigated their anti-MRSA activities againsts-type OM481 and OM584 strains.The results showed that anti-MRSA activity of indoquinoline tetracyclic compounds against MRSA OM481 and OM584 strains was superior to that of tricyclic compound 4-(acridine 9-amino)phenol hydrochloride.The positions and types of substituents in indoquinoline analogues were important for the anti-MRSA activity against both strains.The indoquinoline analogues20a1,which introduced a hydroxyl group at the 4'-position showed obvious anti-MRSA activity against OM481 (MIC=4 μg/mL) and OM584 (MIC=2 μg/mL),revealing that 4'-hydroxyl group was important for the anti-MRSA activity towards both MRSA OM481 and OM584 (Table 6).The introduction of substituents at 7-position of indoquinoline analogues also exhibited good anti-MRSA activity,especially methoxy (20a2)exhibited obvious anti-MRSA activity against both OM481 and OM584 with MIC of 2 μg/mL (Table 6).The anti-MRSA activity of indoquinoline (20a3) and methylindoquinoline (20a4) was weak,but benzofuran-quinoline (20a5) displayed significant anti-MRSA activity against both strains,with an MIC of 2 μg/mL (Table 6).The SARs indicated that the indoquinoline ring,benzofuranquinoline ring,and 4-aminophenol group were the basic structures for the anti-MRSA activity.
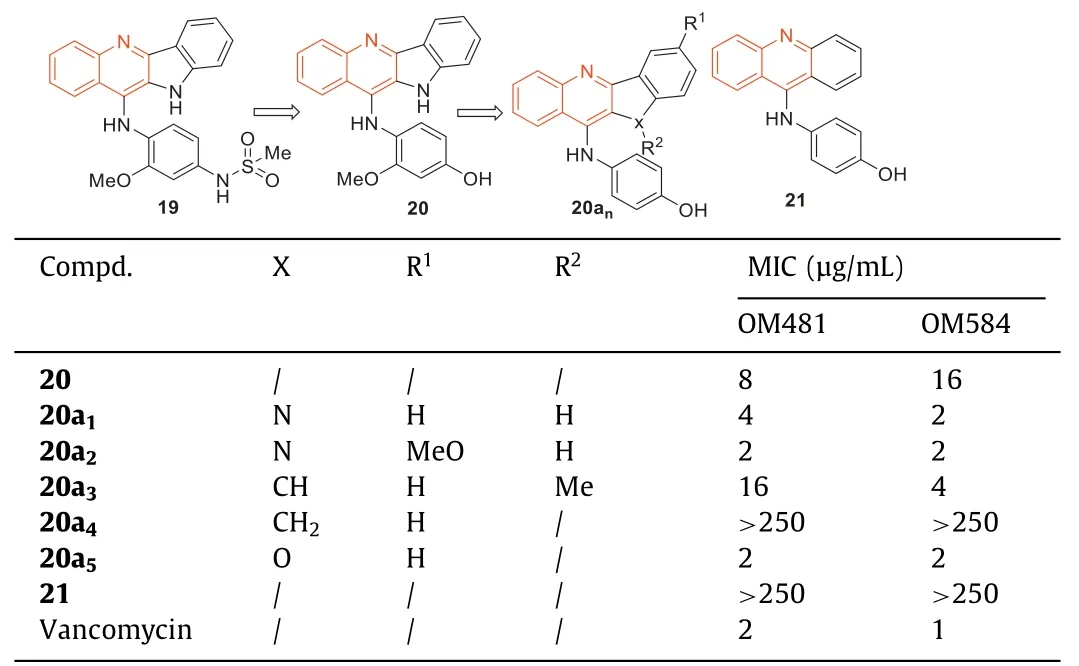
Table 6 Structures of indoquinoline analogues and their anti-MRSA activities.a
5.2-,3-Positions modified quinoline derivatives
In 2016,Shirietal.[77] reported a series of diamide derivatives containing 2-chloroquinoline scaffolds by Ugi condensation reaction of 2-chloroquinoline-3-carboxaldehyde amines,carboxylic acids,and isonitriles.Among them,compound22exhibited the strongest antibacterial activity against MRSA with an MBC value of 0.62 mmol/L,which was stronger than ampicillin but lower than ciprofloxacin (Fig.13).
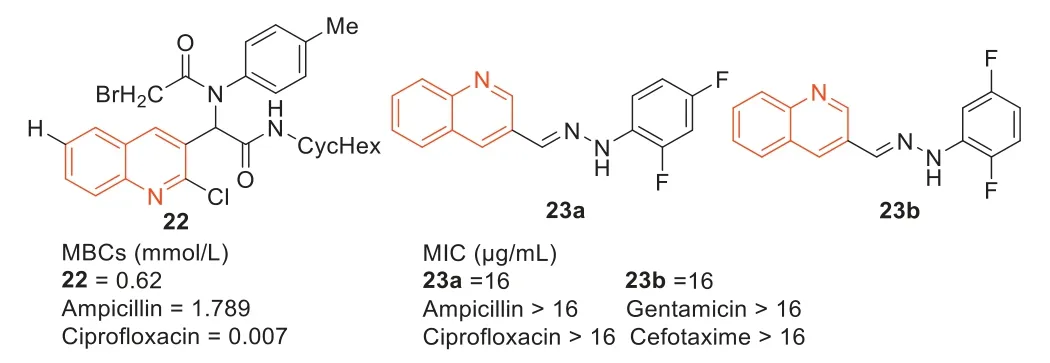
Fig.13. Structure of compounds 22, 23a, 23b and their anti-MRSA activities.
Compounds23aand23bare new compounds synthesized based on quinoline-3-formaldehyde hydrazone compounds (MIC:16 μg/mL),which have a significant antibacterial effect against MRSA compared with ampicillin,gentamicin,ciprofloxacin,and cefotaxime (MIC:>16 μg/mL) (Fig.13).According to the results of MTT test,compounds23aand23bhad no significant effect on McF-7 cells at 100 μmol/L concentration [78].
In 2013,Guoetal.prepared a series of rhodanine derivatives containing the quinoline fraction and evaluated theirinvitroantibacterial activities [79].Theinvitroantibacterial assay showed that most of the rhodanine derivatives exhibited excellent antibacterial activity against different MDR Gram-positive bacteria(MIC value was 1–2 μg/mL).In particular,compounds24aand24b(Fig.14) displayed the strongest level of inhibitory activity(MIC=1 μg/mL),with a 4-8-fold increase in antibacterial activity compared with the standard drug norfloxacin (MIC=4–8 μg/mL).A comparison of carboxylic acid derivatives at theN-position of the rhodanine ring indicated that the contribution order of different carboxylic acids to antibacterial activity was -CH2CH(CH3)2>-CH(CH3)CH2CH3>-CH2C6H5.In addition,the position of substituents on the benzene ring affected the antibacterial activity,and the sequence of antibacterial activity of fluorine-substituted compounds was 4-F>3-F>2-F,and the sequence of antibacterial activity of chlorine-substituted compounds was 4-Cl>3-Cl>2-Cl.Compound24adid not affect the cell viability of human cervical(HeLa) cells when the concentration was equal to the MIC value but showed cytotoxicity at higher concentrations.
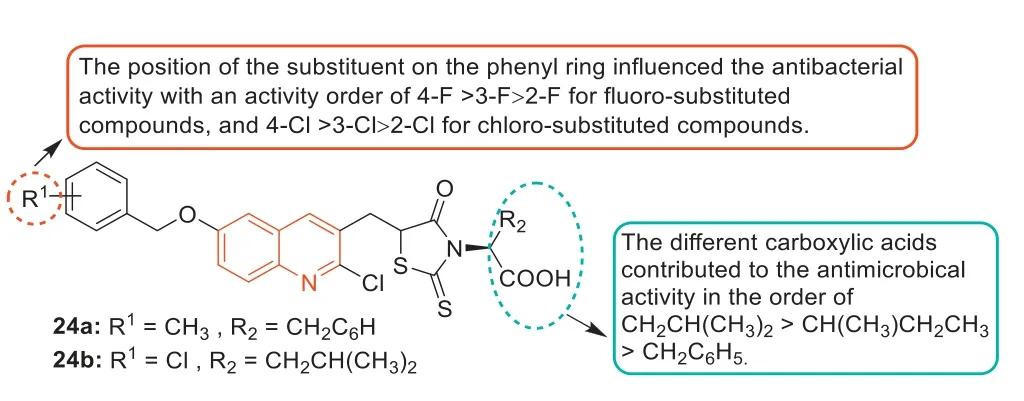
Fig.14. Structures of compounds 24a and 24b and their SARs.
In 2019,Baietal.[80] designed and synthesized some dihydrotriazine derivatives containing the quinoline fraction as novel antimicrobial agents.Dihydrotriazine derivatives25a–c(Fig.15)were the most active compounds against MRSA3506 with an MIC of 1 μg/mL.Based on the activity analysis of these dihydrotriazine derivatives,the following SARs were obtained.The dihydrotriazine group can improve the antibacterial activity of the quinoline compounds.The introduction of phenylethyl at theN-position of the dihydrotriazine ring resulted in significant differences in anti-MRSA activity,suggesting that the substituents of aromatic nuclei were critical for the activity of these dihydrotriazine compounds.In addition,the position of the 2-Cl substituent on the phenyl ring also affected the anti-MRSA activity.The 2,4-di-Cl sub-stituted phenyl ring played a key role in the activity of these dihydrotriazine compounds,and its activity was greater than that of 2,6-Cl substituted compounds.These results were consistent with the previously reported results of a series of rhodanine and dihydrotriazine derivatives [81,11].Invitroenzyme studies showed that compound25aexhibited inhibition of dihydrofolate reductase (DHFR),and compound25adid not show significant cytotoxic activity (HCT116 and LO2 cells IC50>100 μmol/L).These findings suggested that these dihydrotriazine derivatives containing the quinoline fraction could be potential lead compounds for rational development of novel quinoline-based antibacterial agents.
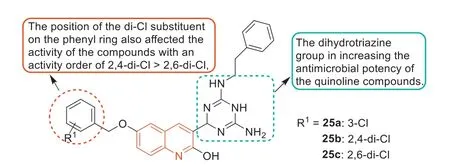
Fig.15. Structures of compounds 25a–c and SARs.
The antimicrobial screening of novel ring-substituted styrylquinolines and two oxorhenium complexes compounds was carried out by Ciesliketal.[82].Compound26(Table 7) was the most effective anti-MRSA candidate,with MIC/IC90values of 3.9 and 7.81 μmol/L at 24 and 48 h,respectively,which were more potent than the standard drugs bacitracin,penicillin V,and ciprofloxacin.
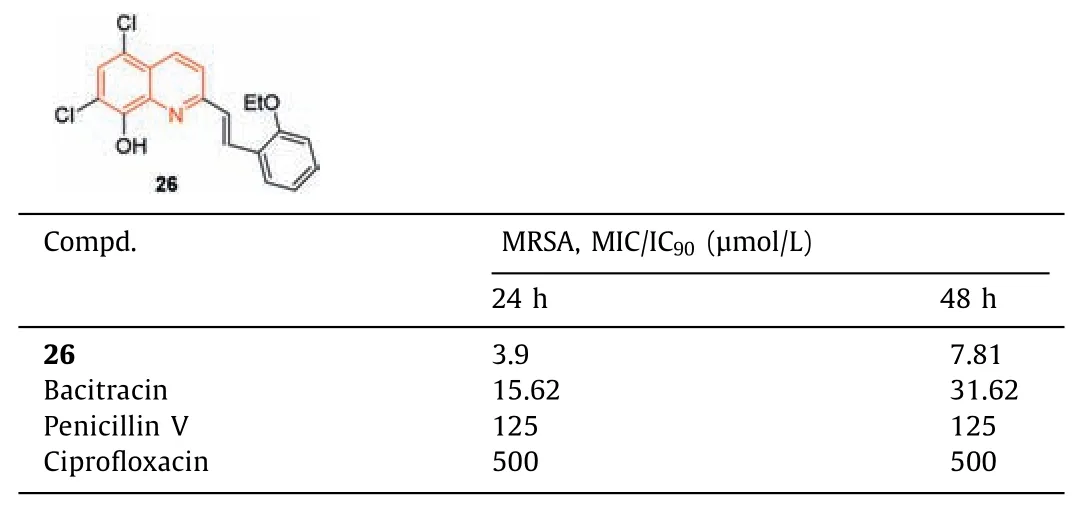
Table 7 Structure of compound 26 and its anti-MRSA activity.
6.4-Position modified quinoline derivatives
In 2018,Fangetal.[83] reported that 1-methylquinolinium iodide derivative (27) had good antibacterial activity against bacterial strains,and it had a synergistic effect in combination withβlactam antibiotics against antibiotic-resistant strains ofS.aureus.The antibacterial activity of27(Table 8,MIC=1.5 μg/mL) against MRSA strains was 100-fold higher than that of methicillin and berberine.Compound27at 0.375 μg/mL significantly increased the antibacterial activity of methicillin against MRSA and decreased the MIC value from 1024 μg/mL to 32 μg/mL.The fractional inhibitoryconcentration index (FICI) of27with methicillin was 0.281.The combination of27with ampicillin or oxacillin also exhibited synergistic effects on MRSA with FICIs of 0.5 and 0.375,respectively.Compound27increased the antibacterial activity of ampicillin by 4 times (MIC decreased from 48 μg/mL to 12 μg/mL) and increased the antibacterial activity of oxacillin against MRSA by 8 times (MIC decreased from 256 μg/mL to 32 μg/mL) at a concentration of 0.375 μg/mL.In addition,when combined with compound27,imipenem and ceftazidime exhibited enhanced antibacterial activity with an FICI of 0.75 (Table 8).Mechanistic studies revealed that compound27inhibited bacterial growth by inhibiting the GTPase activity of FtsZ.
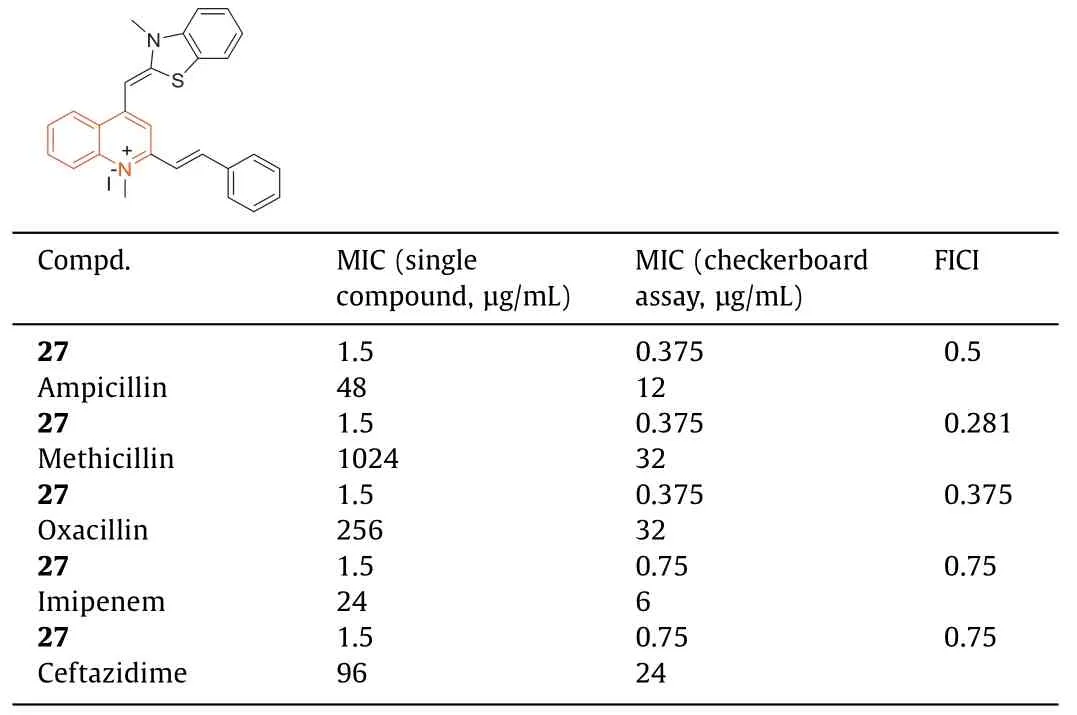
Table 8 MICs of 27 combined with β-lactam antibiotics against drug-resistant S. aureus.a
Tengetal.[84] evaluated the antibacterial activity of eight quinoline derivatives with daptomycin as a reference drug.As shown in Table 9,when both R1and R2groups were kept aspmethyl,the only weak activity of compound28awas detected,and the MIC of28aagainst MRSA was 12 μg/mL.When the R1group was replaced by trifluoromethyl,the resulting compound28bhad better activity with an MIC of 3.0 μg/mL against MRSA.Subsequently,Teng’s group [84] retained the R1as the CF3group and modified R2asm-trifluoromethyl,p-trifluoromethyl,and 3-chloro-4-fluorine groups,respectively,yielding compounds28c,28d,and28e.Compared with28b,quinoline derivative28cshowed no change in antibacterial activity against MRSA,while compound28dexhibited enhanced activity against MRSA with a MIC of 0.75 μg/mL,indicating thep-CF3was more beneficial than thep-CH3group in enhancing the anti-MRSA activity of these quinoline derivatives.Compared with compound28b,the substitution of the 3-chloro-4-fluoro group on compound28ealso showed better activity against MRSA.Based on this result,the R2group of compounds28f,28gand28hwere changed to 3-chloro-4-fluoro groups,but with various R1groups.The quinoline compound28fwith ap-isopropyl phenyl ring also exhibited strong antibacterial activity against MRSA with an MIC value of 1.5 μg/mL.Additionally,compound28gwas synthesized by substituting the phenyl ring in theorthoposition,and it also displayed strong efficacy against MRSA with an MIC value of 1.5 μg/mL.The introduction of an ethoxy spacer between the benzene ring and oxygen at 4-position resulted in a compound28h,whose antibacterial activity was not completely destroyed,with an MIC value of 3.0 μg/mL.
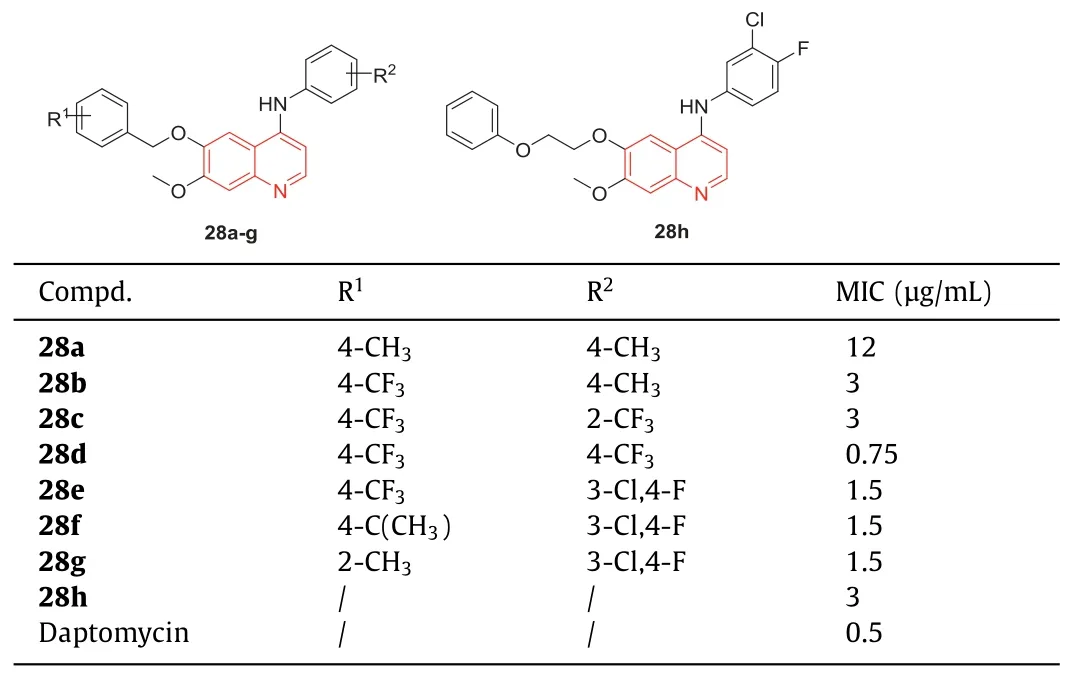
Table 9 Structures of compounds 28a–h and their anti-MRSA activities.
In 2021,compounds29a–r(Fig.16) were synthesized by Manhasetal.[85] using materials reported in the relevant literature to react with various aldehyde condensations.All synthesized quinazolin-4-one Schiff bases29a–rwere subjected toin vitroantibacterial screening,and the zone of inhibition was determined at a concentration of 1.0 mg/mL,and the zone of inhibition between 7 and 9 mm and was considered effective [86].Monofluoroquinazoline-4-ketone was not effective against MRSA,but thep-trifluoromethyl derivative29e,the 3-oxygenated trifluoromethyl derivative29f,the 2-chloro-6-fluoro derivative29g,and the 3,5-difluoro derivative29hexhibited good antibacterial effects against MRSA.Notably,dimethylaminophenyl and 3-hydroxy-4-methoxyphenyl derivatives29kand29lwere also active against MRSA.Therefore,these compounds could be used as lead compounds for modifying their structures and improving their anti-MRSA activities.
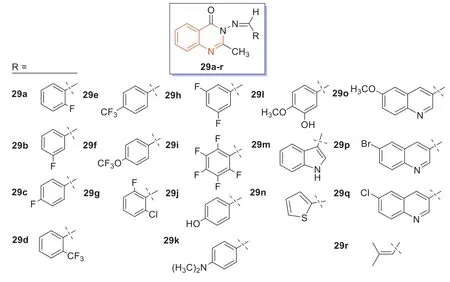
Fig.16. Structures of compounds 29a–r.
During 2000–2016,Dinakaranetal.,Strigacovaetal.,and Wangetal.[87–89] synthesized three series of quinoline-4-carboxylic acid derivatives on the basis of quinoline-4-carboxylic acid and evaluated their antibacterial activities.Among them,compound30a(Fig.17) showed good antibacterial activity against MRSA with an MIC value of 512 μg/mL,which was comparable to the antibacterial activity of ampicillin (Fig.17).Additionally,MTT assay showed that compound30ahad low cytotoxicity.SARs revealed that 2-phenyl ortho amide side chain length had a significant effect on the antibacterial activity,for the same end of the alkaline,with a long side chain (n=2,three bonds between the basic N terminus and carbonyl group) compounds exhibited stronger antibacterial activity than those derivatives with shorter side chain and a carbonyl group (n=1,two bonds between the basic N terminus and carbonyl group),respectively.The cyclic amino group at 2-phenyl group can increase the antibacterial activity of quinoline-4-carboxylic acid derivatives against MRSA.
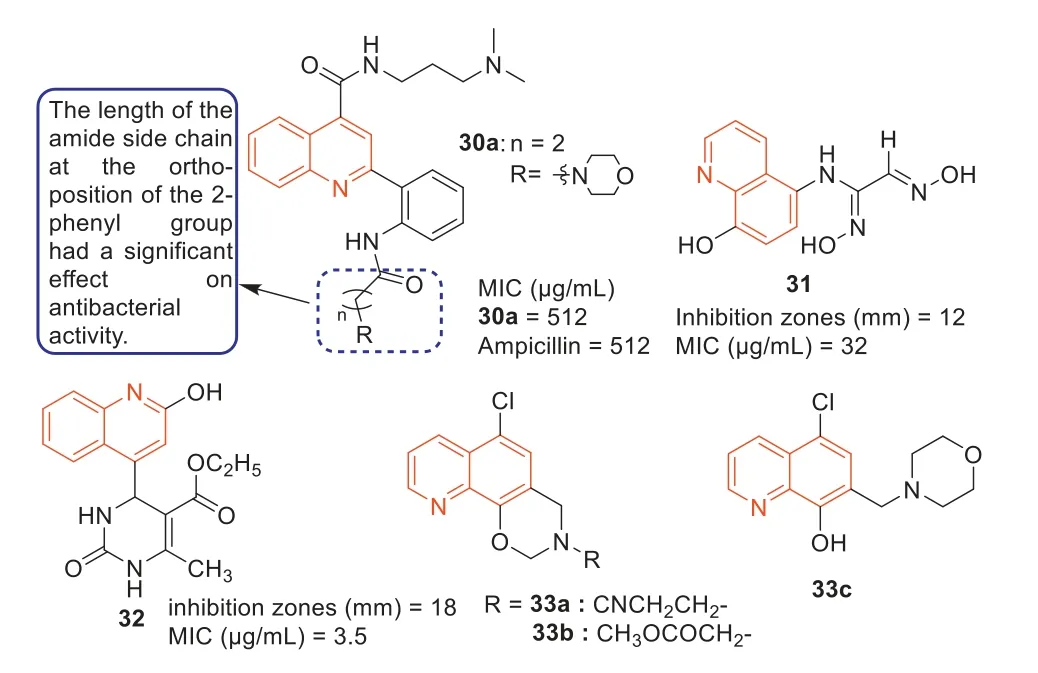
Fig.17. Structure of compounds 30–33 and their anti-MRSA activities.
In 2012,Sevgietal.[90] synthesized several quinolinebased ethylenedione derivatives andN-(8-hydroxyquinolin-5-yl)-aminoethylenedione and evaluated their anti-MRSA activities.It was worth mentioning that compound31(Fig.17) exhibited the most effective anti-MRSA activity with an inhibition zone of 12 mm and an MIC value of 32 μg/mL.Among the dihydropyrimidinone derivatives synthesized by Pauletal.[91],compound32(Fig.17) displayed good antibacterial activity against MRSA with an MIC value of 3.5 μg/mL,superior to the positive control streptomycin (MIC=3.9 μg/mL).
7.7-,8-Positions modified quinoline derivatives
The compound 5-chloro-13-phenethyl-13,14-dihydro-2H-[1,3]oxazino[5,6-H]quinoline (33,Fig.17) was first reported by Enquist’s research group [92],which had a unique scaffold and was effective against Gram-negative bacteria,includingE.coliandPseudomonasaeruginosa.In 2019,Fu’s group firstly elucidated the new mechanism of action of compound33againstE.coli,mainly through blocking lipopolysaccharide (LPS) transport (Lpt)A-LptC interaction by targeting LptA [93].Using compound33as a precursor,a series of oxazole-quinoline derivatives were designed and another series of quinoline derivatives with 5-chloro atoms were constructed,while the hybrid compound33cwas prepared by linking a quinolone fragment as a special substituent to the quinoline core (Fig.17) [94].The antibacterial experiments showed that compounds33a,33b,and33chad potent effects on drugresistant strain MRSA,with MIC values of 8 μg/mL.The results of bacterial molecular docking studies revealed that compound33chad the advantage of dual-targeting mechanism of LptA and topoisomerase IV.
8.Thiourea-containing quinine derivatives
Among the thiourea-containing quinine derivatives (Fig.18)synthesized by Dolanetal.[95],compound34was the most active quinine derivative with an MIC90value of 17.74 μg/mL,which was higher than the reported MIC90value of 1.35 μg/mL or 2 mg/L for vancomycin against MRSA.The SARs in Fig.18 indicated that:(1) loss of the -OMe group can reduce antibacterial potency but it may not be essential for overall activity and loss of the quinoline and -CF3severely reduce anti-MRSA activity;(2) The presence of a sterically bulky group was favorable for anti-MRSA activity and the quinine-derived amine moiety alone was inactive;(3) Structurally simple 3,5-bis(trifluoromethyl)-phenyl thiourea derivatives exhibit little or no anti-MRSA activity.Additionally,toxicity evaluation showed that34was not only non-toxic toGalleriamellonellalarvae but also did not appear to impede larvae development.Furthermore,compound34was also non-toxic toG.mellonellalarvae at concentrations up to 1000 μg/mL.Encouraged by these results,thiourea-containing quinine derivative34had great potential as a new antibacterial agent against MRSA.
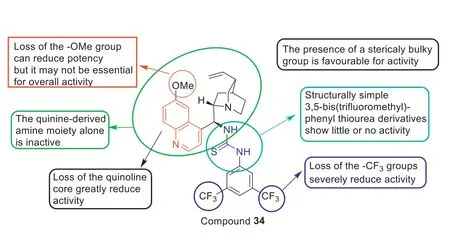
Fig.18. SARs of quinine-based thiourea 34.Reproduced with permission [95],Copyright 2016,Elsevier publications.
9.Antibacterial mechanism of quinoline analogues against MRSA
Here,we briefly summarized the anti-MRSA mechanisms of some representative quinoline analogues based on the types of targets and the process of action between quinoline analogues and bacteria,mainly including the interaction of DNA topoisomerase,disruption of cell membranes and inhibition of the FtsZ associated with cell division.
9.1. DNA topoisomerase
It is well known that DNA topoisomerases are the main targets of quinolone antibiotics.Herein,compound3dwas able to inhibit the relaxation activity of EcTopo IV at a concentration of 10 μmol/L[40].Interactions of compound3d(Fig.19A) with DNA topoisomerase IV receptor were shown in Figs.19B and C,the carboxyl group of this molecule was very close to residue Arg117 of the DNA topoisomerase IV complex.Compound3dcan also form hydrogen bonds with Ser79 of the DNA topoisomerase IV complexviathe hydrogen atom of the carboxyl group.In addition,compound3dcan insert into the superhelical DNA of the enzyme-DNA complex.This synergistic binding may facilitate the formation of a stable quinolone-DNA enzyme complex,which may account for the strong inhibitory effect of3don MRSA.Moreover,Zhangetal.[40] used propidium iodide (PI) to study the ability of3dto penetrate bacterial cell membranes and found that3dcan cross the damaged bacterial cell membranes and bind to DNA to emit stronger fluorescence (Fig.19D).
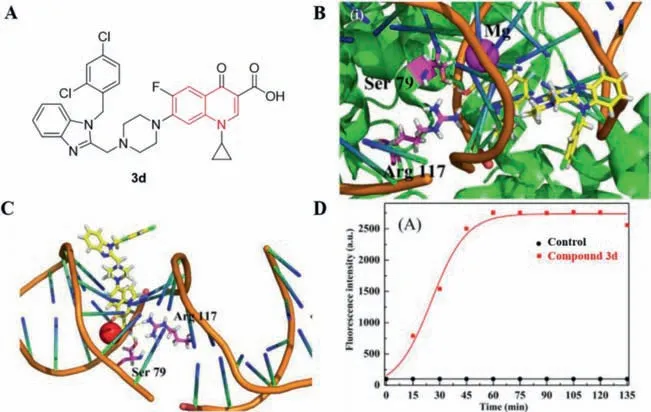
Fig.19. (A) Structure of 3d;(B,C) Three-dimensional conformation of 3d docked in topoisomerase IV–DNA complex;(D) Mechanism of antibacterial action of 3d at concentrations of 12×MIC.Reproduced with permission [40],Copyright 2016,Elsevier publications.
9.2. Cell membranes
The bacterial cell membrane is one of the important structures on which bacteria depend for survival and is rich in enzyme systems that perform many important metabolic functions.In 2010,Huetal.reported that a quinoline derivative HT61 (1)can act on the plasma membrane of bacterial cells by disrupting the bacterial cell membrane potential of MSSA,resulting in the release of cellular contents [38].The polarization effect of the quinoline derivative HT61 on MSSA bacterial membranes was detected by the fluorescent probe 3,3-dipropylthiadicarbocyanine iodide (DiSC3(5)).As shown in Fig.S1A (Supporting information),1 min after the addition of HT61,the fluorescence intensity was enhanced in a concentration-dependent manner for the stationary phase MSSA.While the fluorescence intensity did not increase significantly after treatment for 1 min.This result suggested that the maximum depolarization of the cytoplasmic membrane of stationary phase MSSA occurred within 1 min to 4 min after being treated with HT61.However,for log phase MSSA (Fig.S1B,Supporting information),the release of fluorescence after HT61 treatment was slower than that of the stationary phase and achieved a peak after 45 min.It appears that the increased fluorescence intensity was concentration-dependent only at lower concentrations of HT61.When HT61 reached higher concentrations,the fluorescence release did not increase when the concentration of HT61 was higher than 20 mg/mL.Furthermore,transmission electron micrographs showed that after treatment with HT61 for 10 min,the cell membrane ofS.aureuswas disrupted and the cytoplasm exuded into the extracellular space (Fig.S2B,Supporting information).Additionally,after treatment with HT61 at higher concentrations,the cell wall cracked and the cell contents were expelled (Figs.S2C and D,Supporting information).
9.3. FtsZ
FtsZ is highly conserved in bacteria and is essential for cell division [96].Recent studies have reported that quinoline derivatives induce cell death in antibacterial assays possibly due to their inhibitory effect on the GTPase activity of FtsZ,making FtsZ a novel target for the development of broad-spectrum antibiotics [97].As shown in Fig.S3 (Supporting information),compound17b2strongly inhibited Sa-FtsZ polymerization in a dose-dependent manner,exhibiting a significant inhibition of FtsZ polymerization at a concentration of 16 μg/mL [65].The effect of theN-methylbenzindolo[3,2-b]quinoline derivative17b2on FtsZ polymerization was also analyzed by transmission electron microscopy by Sunetal.[65].As displayed in Fig.S4A (Supporting information),a dense network of FtsZ protofilaments with an average width of 104 ± 18 nm was observed in the absence of17b2.While a concentration of 8 μg/mL of17b2greatly reduced the size and thickness of FtsZ polymer and the bundling of FtsZ protofilaments(Fig.S4B,Supporting information).Since the GTPase activity of FtsZ correlates with the dynamic polymerization of FtsZ [98],the effect of compound17b2on the dynamic polymerization of FtsZ was investigated.In the GTPase assay,17b2showed about 20% inhibition at a concentration of 4 μg/mL (Fig.S4C,Supporting information).However,approximately 50% and 60% inhibition were reached at concentrations of 8 and 16 μg/mL,respectively.These results suggested that17b2significantly inhibited the GTPase activity of FtsZ in a dose-dependent manner.In 2018,Fangetal.[83] found that compound27inhibited bacterial growth by inhibiting the GTPase activity of FtsZ.They used transmission electron microscopy to find a significant increase in the size of the FtsZ polymer after incubation of FtsZ with27at a concentration of 1.5 mg/mL (Figs.S4D and E,Supporting information).Moreover,compound27at 0.75 mg/mL showed about 20% inhibitory activity,and27at concentrations of 1.5,3,and 6 mg/mL can achieve 50%,70%,and 80%inhibition effects (Fig.S4F,Supporting information).These results suggested that the anti-MRSA activity of compounds17b2and27may be due to their inhibitory effects on GTPase activity and FtsZ polymerization.
9.4. DHFR
DHFR,is involved in the biosynthetic pathway of folate,a nitrogenous base and precursor of some amino acids.DHFR is one of the key components of this pathway and inhibition of DHFR can be lethal to bacteria.Thus,DHFR can be used as a target against pathogenic bacteria.Baietal.[80] reported that compound25awas well inserted into the active pocket ofS.aureusDHFR(Fig.20A) and it could reduce the activity of DHFR by 81% at a concentration of 50 μmol/L compared to the negative control(Fig.20B).It was confirmed that compound25ashowed an inhibitory effect on DHFR,indicating that DHFR may be a potential antibacterial target [80].
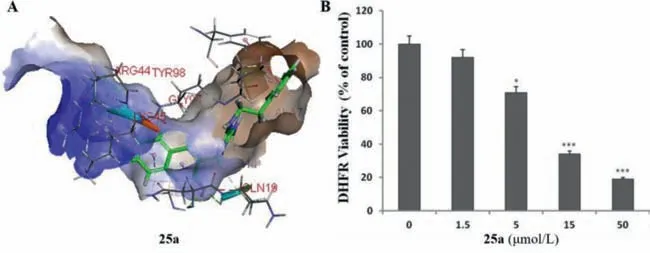
Fig.20. Inhibition of DHFR activities of compound 25a.Reproduced with permission [80],Copyright 2019,John Wiley publications.∗P <0.05,∗∗∗P <0.001,significant with respect to the control (0 μmol/L) group.
10.Conclusions and outlook
In this review,we addressed what is known about the antimicrobial activity of quinoline analogues over the past two decades,particularly against MRSA,their SARs,as well as mechanisms.Quinoline analogues have a wide range of biological and pharmaceutical activities,and a number of quinoline analogues have been designed,developed,and screened against Gram-positive,Gramnegative,and MDR bacteria in recent two decades.Many quinoline analogues exhibited more potent antibacterial activity against MRSA than the reference antibiotics and were less toxic.The SARs suggested that the anti-MRSA activity of quinoline analogues is related to the position,species,and spatial relationships of the various substituents.Their abundant SARs may provide better insights for further rational development of quinoline-based antimicrobials to combat MRSA infections.Additionally,the elucidation of the anti-MRSA mechanism of some representative quinoline analogues will provide a reference for the design of novel quinoline-based antibacterial drugs in the future.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
We thank the National Natural Science Foundation of China(No.32272575) and National College Student Innovation and Entrepreneurship Training Program (No.202210459164) for financial support.
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.cclet.2023.108511.
 Chinese Chemical Letters2023年12期
Chinese Chemical Letters2023年12期
- Chinese Chemical Letters的其它文章
- Spin switching in corrole radical complex
- Benzothiadiazole-based materials for organic solar cells
- Mono-functionalized pillar[n]arenes: Syntheses,host–guest properties and applications✰
- Recent advances in two-step energy transfer light-harvesting systems driven by non-covalent self-assembly✩
- From oxygenated monomers to well-defined low-carbon polymers
- Doping-induced charge transfer in conductive polymers