
Design and synthesis of tri-substituted pyrimidine derivatives as bifunctional tumor immunotherapeutic agents targeting both A2A adenosine receptors and histone deacetylases
2023-02-18 01:55RuiqunLiuWenwenDunWenzhongYnJinfengZhngJinjunCheng
Chinese Chemical Letters 2023年12期
Ruiqun Liu ,Wenwen Dun ,Wenzhong Yn ,Jinfeng Zhng ,Jinjun Cheng,b,∗
a iHuman Institute,ShanghaiTech University,Shanghai 201210,China
b School of Life Science and Technology,ShanghaiTech University,Shanghai 201210,China
Keywords:Bifunctional A2AAR antagonism HDAC inhibition Cancer Immunotherapy
ABSTRACT The A2A adenosine receptor (A2AAR) has attracted attention as an emerging immunotherapeutic target with several antagonists being evaluated in clinical trials.However,A2AAR antagonists show limited efficacy as monotherapies.Herein,we communicate our design and synthesis of a novel series of A2AAR/histone deacetylase (HDAC) bifunctional inhibitors,based on the core structure of the A2AAR antagonist PBF-509.The new compounds were designed using a pharmacophore-merging strategy and features a tri-substituted pyrimidine core.The binding affinity for A2AAR and inhibitory activity against HDACs of all the new compounds were tested.A number of compounds exhibited nanomolar or subnanomolar activity against both targets and some showed equally potent antiproliferative activity against MC38,CT26 and HCT116 colon cancer lines compared to HDAC inhibitors SAHA and MGCD-0103 in vitro.The binding poses of compound 5a in both A2AAR and HDAC1 were predicted by molecular docking studies.Collectively,these results suggest these tri-substituted pyrimidine derivatives are promising leads for developing A2AAR/HDAC dual-acting compounds as novel antitumor agents.
The development of immunotherapy has been a milestone for the treatment of cancer.Successful immunotherapeutics include checkpoint inhibitors targeting the programmed death-1 (PD-1),programmed death-ligand 1 (PD-L1) and cytotoxic T lymphocyteassociated antigen-4 (CTLA-4).However,tumor has immune escaping mechanisms and the efficacy of existing strategy is limited.Therefore,the development of novel immunotherapeutics targeting alternative immune escaping pathways is urgent.
In the tumor microenvironment (TME),the critical mechanism of tumor immune escape has been shown to be mediated by the accumulation of extracellular adenosine [1,2].Among the four subtypes of adenosine receptors,the A2AAR has been demonstrated as the major target that mediates the immunosuppressive effects of adenosine in TME,which is highly expressed on multiple T cells and natural killer cells [3–6].Recently,a number of A2AAR antagonists have been developed and are undergoing extensive evaluations in clinical trials as potential cancer immunotherapies (e.g.PBF-509) (Fig.1) [7,8].However,A2AAR antagonists show limited antitumor effects and are more commonly tested in combination with cytotoxic drugs or other checkpoint inhibitors [7].From a medicinal chemistry point of view,the design of polypharmacological molecules would be a better alternative to drug combinations,with advantages such as the absence of drug-drug interactions and better patient compliance [9].
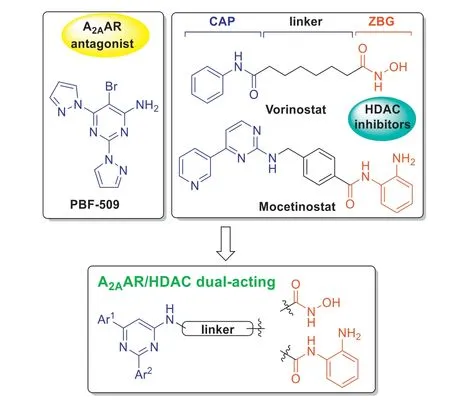
Fig.1. Structures of the A2AAR antagonist PBF-509,HDAC inhibitors vorinostat and mocetinostat,and the design of dual A2AAR/HDAC inhibitors.
The histone deacetylases (HDACs) are validated epigenetic drug targets,and several HDAC inhibitors have been developed as antitumor drugs.Chemical structures of HDAC inhibitors (HDACis),for example vorinostat (SAHA) and mocetinostat (MGCD-0103),are commonly comprised of three parts: the surface recognition moiety (CAP),a linker group,and the terminal zinc-binding group(ZBG) (Fig.1).These structural features have made HDACs a popular target for the design of bifunctional antitumor agents.For example,DNMT/HDAC [10,11],EGFR/HDAC [12,13],ErbB/HDAC [14],PI3K/HDAC [15],topisomerase/HDAC [16,17],and JAK/HDAC [18,19],have been reported.In our previous studies,a bifunctional strategy has been used for the design of novel A2AAR/HDAC dual-acting agents,which led to the discovery of potent antitumor agents[20,21].Recently,based on the clinical agentPBF-509,we have initiated another campaign to design novel A2AAR/HDAC dual-acting compounds (Fig.1).
Among the clinically investigated A2AAR antagonists,PBF-509features a relatively low molecular weight (Mw=305).To rationally design bifunctional compounds based onPBF-509,we first conducted a molecular docking study using the reported A2AARStaR2-bRIL562-AZD-4635 complex structure (PDB ID: 6GT3) [22].As shown in Fig.2,the 2-pyrazole substituent ofPBF-509occupies a hydrophobic cleft at the bottom of the binding pocket.The 6-pyrazole substituent occupies the ribose-binding pocket.The NH2group makes an H-bond interaction with the side chain of Asn2536.55.In addition,Phe168 on extracellular loop 2 (ECL2)formsπ-πstacking interactions with the pyrimidine and 6-pyrazole,and the side chain of Met2707.35makes a hydrophobic interaction from the opposite side.The NH2group ofPBF-509points towards the solvent accessible surface,therefore providing a modification site for incorporating structural elements of HDACis.Based on this NH2group,three types of linker groups were designed:amide,urea and carbamate.
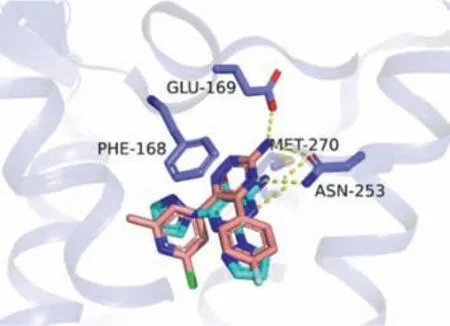
Fig.2. Binding poses of PBF-509 in A2AAR predicted by molecular docking.Structure of A2AAR-StaR2-bRIL562 (PDB ID: 6GT3) is shown in pale gray, AZD-4635 in salmon,and compound PBF-509 in cyan,respectively.Key residues interacting with PBF-509 are highlighted.Nitrogen atoms are colored in blue,bromine in firebrick,fluorine in aquamarine and chlorine in green.
PBF-509was prepared according to reported procedures [23],but coupling reactions of it with aliphatic acids failed to provide amide intermediates.So the bromo group was removed.Variations of the aromatic substituents on the pyrimidine core were incorporated,based on the results from reported structure-activity relationship studies [24–26].As shown in Scheme 1 (details can be found in Supporting information),starting materials1a-dwere coupled with a variety of aliphatic acids to produce the key ester intermediates2a-g.However,cleavage of the amide bond wasobserved when these ester intermediates were treated with aqueous hydroxylamine under basic condition.Therefore,the esters2agwere first hydrolyzed to acids3a-gin the presence of LiI and pyridine [27].Then3a-gwere coupled withO-(tetrahydropyran-2-yl)hydroxylamine to give4a-g,and hydroxamic acids5a-gwere obtained after de-protection of the pyran group (Scheme 1,I).
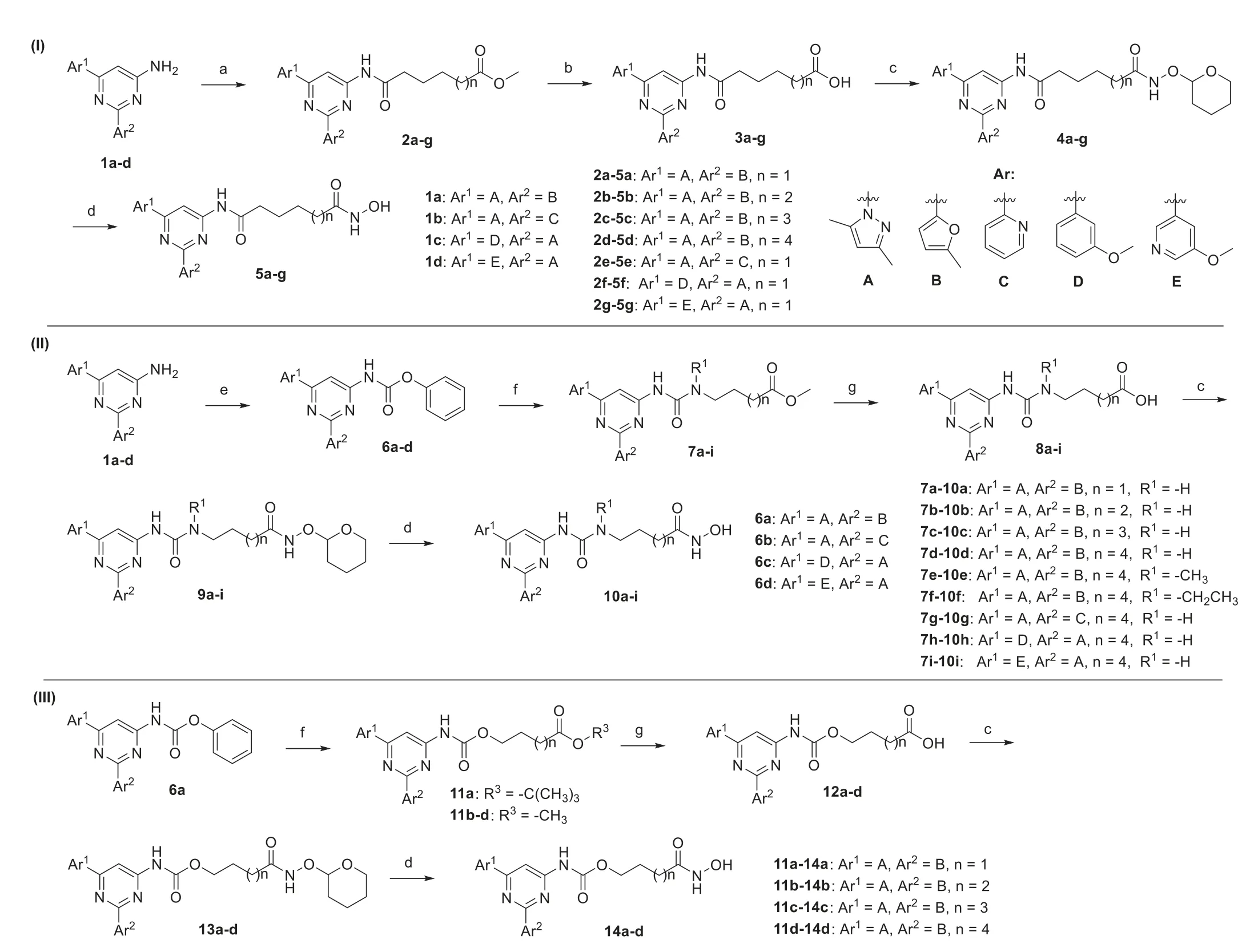
Scheme 1. Synthesis of compounds 5a-g, 10a-i and 14a-d.Reagents and conditions: (a) (i) SOCl2,acid esters,benzotriazole,DCM,r.t.,1 h;(ii) pyridine,DCM,r.t.,12 h;(b)LiI,pyridine,reflux,12 h;(c) 2-(aminooxy)tetrahydro-2H-pyran,HATU,DIPEA,DCM,r.t.,12 h;(d) HCl (4 mol/L in 1,4-dioxane),DCM,0 °C– r.t.,2 h;(e) phenyl chloroformate,pyridine,DCM,r.t.,12 h;(f) hydroxyl esters or amino esters,DIPEA,THF,CHCl3,reflux,12 h;(g) LiOH·H2O,THF,H2O,r.t.,12 h.
To synthesize urea-and carbamate-linked compounds,the materials1a-dwere first coupled with phenyl chloroformate to provide intermediates6a-d,then substituted with a variety of amines or alcohols to provide the key ureas7a-iand carbamates11a-d.These intermediates were then hydrolyzed to provide acids8aiand12a-d,coupled withO-(tetrahydropyran-2-yl)hydroxylamine,and then hydrolyzed to afford the corresponding hydroxamic acids10a-iand14a-d(Scheme 1,II and III).
A combination of an urea group and a benzyl linker was also introduced,and the compounds were synthesized according to Scheme 2 (details can be found in Supporting information).The carbamates6a-dwere converted to ester intermediates15a-eand then hydrolyzed to acids16a-e.These acids were converted to hydroxamates18a-eusing similar conditions as described above.Acids16a-dwere also coupled witho-phenylenediamine to afford amide compounds19a-d.
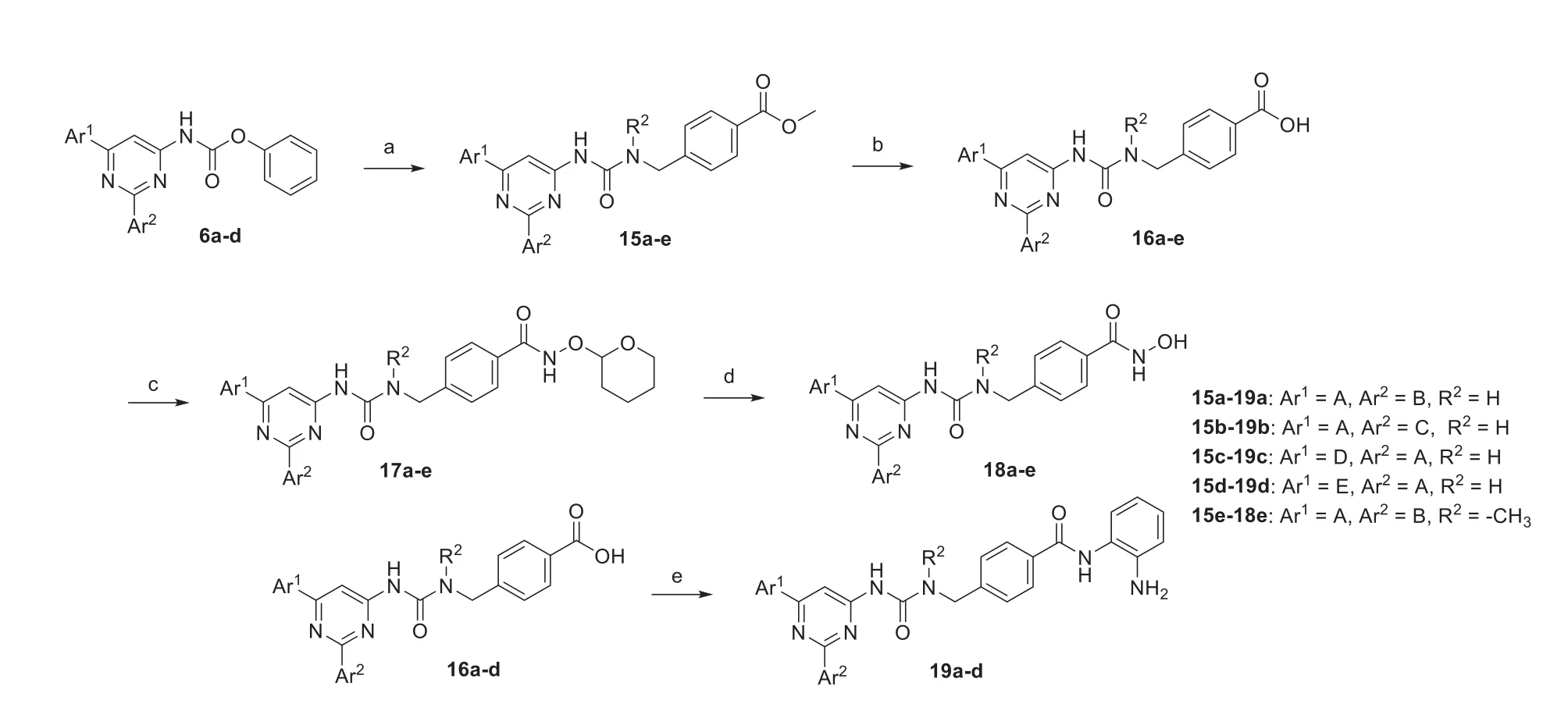
Scheme 2. Synthesis of compounds 18a-e and 19a-d.Reagents and conditions: (a) hydroxyl esters or amino esters,DIPEA,THF,CHCl3,reflux,12 h;(b) LiOH·H2O,THF,H2O,r.t.,12 h;(c) 2-(aminooxy)tetrahydro-2H-pyran,HATU,DIPEA,DCM,r.t.,12 h;(d) HCl (4 mol/L in 1,4-dioxane),DCM,0 °C–r.t.,2 h;(e) o-phenylenediamine,HATU,DIPEA,DCM,r.t.,12 h.
As shown in Table 1,most target compounds displayed potent dual-acting activities,with potent A2AAR binding affinity (PBF-509as a positive control [28]) and HDAC1/HDAC6 inhibition (SAHAas a positive control),respectively.Among the three types of linker groups,amide connection provided the best activity (10dversus5c,14d).Regarding the length of the alkyl chain,5c,10dand14cwere the best with 6 or 5 carbon chains,while longer or shorter chains showed weaker activity (5cversus5a,5b,5d;10dversus10a,10b,10c;14cversus14a,14b,14d).For ureas,alkyl substitution weakened the activity (10dversus10e,10f),and the same was true for aromatic linkers (18aversus18e).For the ZBG group,compounds with 2-amino benzamide as the ZBG showed weaker HDAC activity than the hydroxamic acids.Finally,the furyl and pyrazol substituents on the pyrimidine core were replaced with pyridyl and phenyl groups,and appropriate linkers were incorporated to synthesize compounds10g-i,18b-d,and19b-d.The activity of these pyridine and 2-methoxypyridine-containing compounds was slightly better.

Table 1 A2AAR and HDAC bifunctional activity of designed compounds.a
The compounds with potent bifunctional activities were tested for their antiproliferative activities against both murine and human colon cancer cell lines (MC38,CT26 and HCT116).The results are shown in Table 2.Because A2AAR antagonists do not possess direct cytotoxicity against the tumor cellsinvitro[29,30],the antiproliferative activity of these compounds were expected to be correlative to their HDAC inhibition potency.In these assays,compoundPBF-509showed no observable activity (GI50>30 μmol/L).Among the dual-acting compounds,5a,10c,10dand18adisplayed the most potent activity against the three cell lines,being comparable to that ofSAHA(GI50=2.7 μmol/L,2.4 μmol/L and 0.30 μmol/L for MC38,CT26 and HCT116,respectively) andMGCD-0103(GI50=1.8 μmol/L,2.1 μmol/L and 0.53 μmol/L for MC38,CT26 and HCT116,respectively).These results showed that through the incorporation of HDAC inhibition activity,the bifunctional compounds acquired very potent antiproliferative activity against tumor cellsinvitro.It worth pointing out that some compounds,for example18d,showed very weak antiproliferative activity despite very potent HDAC inhibition.This might be due to their poor permeability properties.
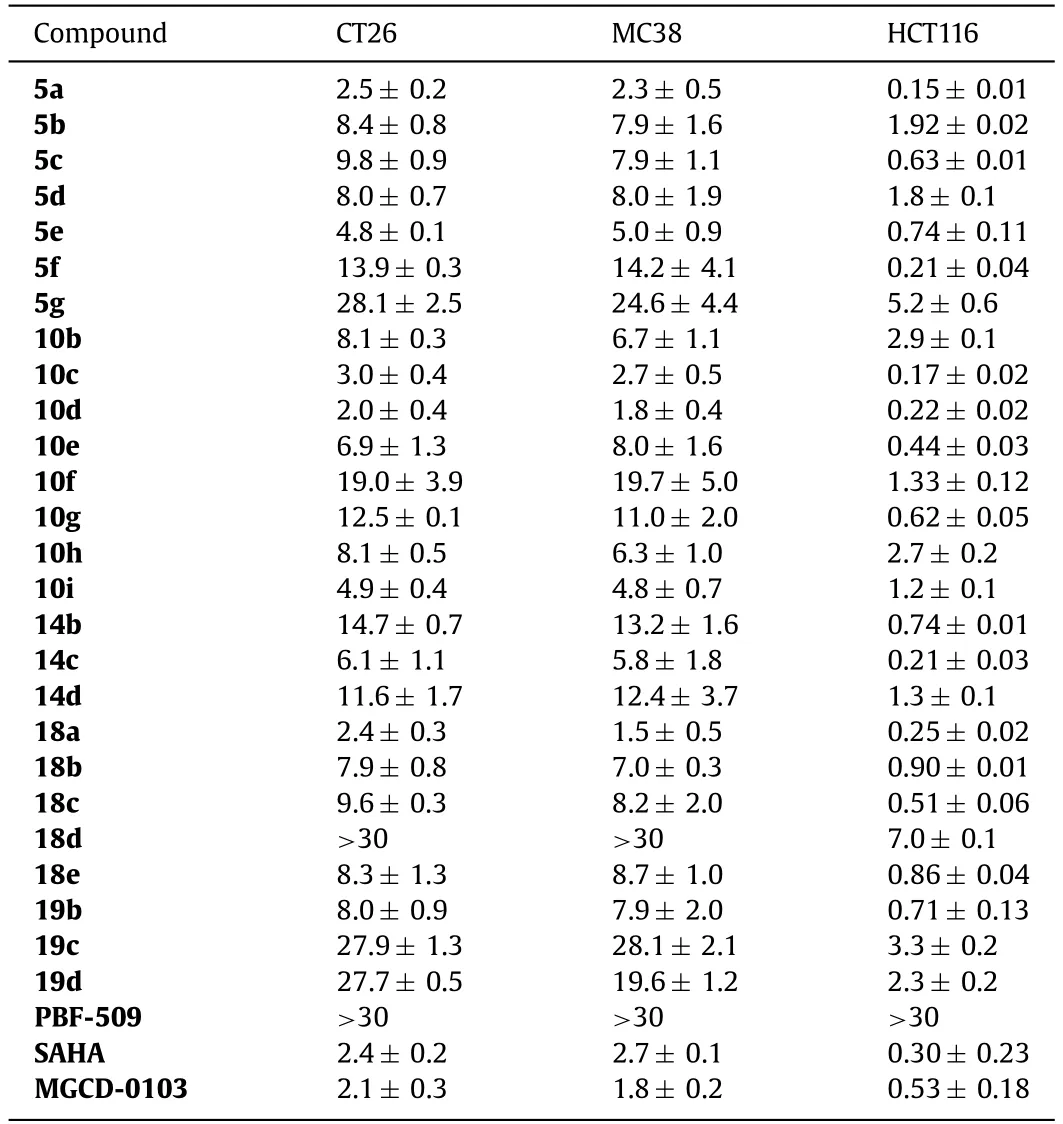
Table 2 Antiproliferative activity in cancer cell lines (GI50,μmol/L).a
To predict the binding modes of these bifunctional molecules at both A2AAR and HDACs,compound5awas selected for molecular docking studies (Fig.3).The 2-methylfuran substituent of5aoccupies the hydrophobic pocket,with the dimethylpyrazole substituent fitting into the ribose-binding pocket deeper inside the receptor.In addition,Phe168 on ECL2 forms aπ-πstacking interaction with the pyrimidine moiety.Unexpectedly,the hydroxamate group makes H-bonding interactions with the side chain of Asp170 and the skeleton of Glu169.In HDAC1,the core structure of5alies outside of the binding pocket,with the alkyl chain inserting into the pocket and the hydroxamate group chelating with the zinc ion.The carbonyl group of the amide linker makes an H-bond with the side chain of His170.The hydroxamate group also makes H-bond interactions with the side chains of His131 and Gly295.The trisubstituted pyrimidine core lies on the enzyme surface and forms an edge-to-faceπ-πinteraction with Phe200.These observations predicted the structural basis of the bifunctional activity of our new compounds.
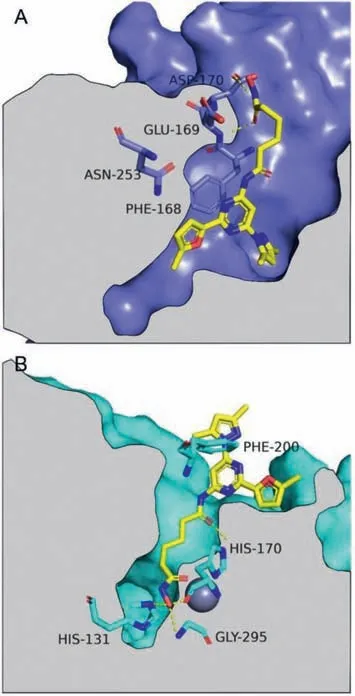
Fig.3. Binding poses of 5a in A2AAR and HDAC1 predicted by molecular docking.Compound 5a is shown in yellow.Key residues interacting with 5a are highlighted.Nitrogen atoms are colored in blue and oxygen atoms in red.(A) The predicted binding pose of 5a in A2AAR.Structures of A2AAR-StaR2-bRIL562 (PDB ID: 6GT3) is shown in deepblue.(B) The predicted binding pose of 5a in HDAC1.Structures of HDAC1 (PDB ID: 1C3S) is shown in cyan,and the zinc ion is shown as gray sphere.
In summary,the incorporation of the key structural element for HDAC inhibition to the solvent-exposed position of A2AAR has culminated the discovery of a series of potentPBF-509-derived A2AAR/HDAC bifunctional inhibitors.Most compounds displayed nanomolar or subnanomolar inhibitory activity against both A2AAR and HDAC1.In addition,they exhibited equivalent antiproliferative activity to that ofSAHAandMGCD-0103against colon cancer cell lines.Based on the A2AAR/HDAC bifunctional activity of these compounds,they are promising leads for further studies as novel tumor immunotherapeutic agents.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported by Lingang Laboratory (No.LG-QS-202205–03),ShanghaiTech University and the Shanghai Municipal Government.We acknowledge Dr.Lingyun Yang for his help in obtaining NMR data.
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.cclet.2023.108136.
 Chinese Chemical Letters2023年12期
Chinese Chemical Letters2023年12期
- Chinese Chemical Letters的其它文章
- Spin switching in corrole radical complex
- Benzothiadiazole-based materials for organic solar cells
- Mono-functionalized pillar[n]arenes: Syntheses,host–guest properties and applications✰
- Recent advances in two-step energy transfer light-harvesting systems driven by non-covalent self-assembly✩
- From oxygenated monomers to well-defined low-carbon polymers
- Doping-induced charge transfer in conductive polymers