
Partial oxidation of methane by photocatalysis
2023-02-18 01:54ZhongshnYngQiqiZhngHuiSongXinChenJiweiCuiYnhuiSunLequnLiuJinhuYe
Chinese Chemical Letters 2023年12期
Zhongshn Yng ,Qiqi Zhng ,Hui Song ,Xin Chen ,Jiwei Cui ,Ynhui Sun ,Lequn Liu,∗,Jinhu Ye,
a TJU-NIMS International Collaboration Laboratory,School of Materials Science and Engineering,Tianjin University,Tianjin 300072,China
b International Center for Materials Nanoarchitectonics (WPI-MANA),National Institute for Materials Science (NIMS),1-1 Namiki,Ibaraki 3050047,Japan
c Zhejiang LAMP Co.,Ltd.,Wenzhou 325206,China
Keywords:Partial oxidation of methane Photocatalysis Liquid oxygenates Oxidants C-H activation
ABSTRACT Methane chemistry is one of the “Holy Grails of catalysis”.It is highly desirable but challenge to transform methane into value-added chemicals,because of its high C-H bonding energy (435 kJ/mol),lack of π bonding or unpaired electrons.Currently,commercial methane conversion is usually carried out in harsh conditions with enormous energy input.Photocatalytic partial oxidation of methane to liquid oxygenates(PPOMO) is a future-oriented technology towards realizing high efficiency and high selectivity under mild conditions.The selection of oxidant is crucial to the PPOMO performance.Hence,attentions are paid to the research progress of PPOMO with various oxidants (O2,H2O,H2O2 and other oxidants).Moreover,the activation of the selected oxidants is also highly emphasized.Meanwhile,we summarized the methane activation mechanisms focusing on the C-H bond that was broken mainly by •OH radical,O−specie or photogenerated hole (h+).Finally,the challenges and prospects in this subject are briefly discussed.
1.Introduction
Methane,a feedstock for chemicals and the stable structure of regular tetrahedron,its efficient conversion has attracted growing attention from scientific community and industrial circles [1–3].It is highly desirable but challenge to transform methane into valuable chemicals,because of its high C-H bonding energy (435 kJ/mol),lack ofπbonding or unpaired electrons.Hence,the effi-cient conversion of methane into value-added liquid oxygenates is regarded as a “holy grail” or “dream reaction” in catalysis.
The conversion and utilization of methane mainly rely on two routes [4,5].One is indirect conversion route,or syngas route,which is the current commercial route for large-scale transformation of methane [6–8].This indirect route traditionally takes place under harsh conditions (high temperature or high pressure)with tremendous energy consumption,which exerts great pressure on sustainable development.The other is direct conversion route,in which methane could be transformed directly into liquid oxygenates,syngas,hydrocarbons [9–11].Among various direct conversion routes,partial oxidation of methane (POM) is one of the most attractive routes due to the less energy input and atomic economy.Compared with partial oxidation methane to syngas,the direct conversion of methane to liquid oxygenates is more attractive and challenging for carbon neutralization and sustainable development.The research of POM to liquid oxygenates (POMO) can be traced back to the early 1970s,and Shilov found that methane could be oxidized into methanol with the K2PtCl6as an oxidant at relatively low temperature (120 °C) [12].Although much progress has been made to transform methane into value-added liquid oxygenates since then,it still suffers from extensive energy consumption [13–20].However,the methane conversion within-situgenerated H2O2maybe bring new inspiration [18].
Photocatalysis,as a burgeoning technology,has recently attracted enormous attention owing to it could directly convert solar energy into chemical energy under mild conditions through water splitting,carbon dioxide reduction,environmental remediation and organic transformation [21–58].It is therefore extremely attractive and meaningful to achieve efficient partial oxidation of methane into liquid oxygenatesviaphotocatalysis.Photocatalytic partial oxidation of methane to liquid oxygenates (PPOMO) has been paid increasing attention since Kaliaguine firstly reported the CH4activation to CH3O−species by hole centers (O−) over TiO2under UV irradiation in 1978 [59].CH3OH [60],HCHO [61–67],HCOOH [68] were subsequently reported as main products from CH4over series of metal oxides under light irradiation.These pioneer works primarily validate the feasibility of photocatalytic direct valorization of methane to value-added liquid oxygenates.However,the PPOMO process is usually accompanied by products overoxidation to carbon oxides (CO,CO2),which would lead to a low liquid oxygenates selectivity.Hence,it is challenging and significant to regulate the reaction systems,such as selection of oxidants,design of photocatalysts,reaction temperature/pressure/time and light source.Recent years,quite a lot of progresses on catalytic CH4conversion have been reviewed [69–80].Ordomsky summarized the major routes in the photocatalytic methane conversion into chemicals and fuels under mild conditions [81].The design of efficient photocatalysts for methane conversion was reviewed [82–85].Most of present review focus either on the thermo/electro/photochemical routes of CH4utilization or on material design,while the attention paid on the selection of oxidants and reaction route are limited.Oxidants selection is of great importance to product selectivity as well as the overoxidation regulation.Hence,an updated,profound and thorough review on partial oxidation of CH4into liquid oxygenatesviaphotocatalysis from the perspective of oxidants selection and activation is of great significance and necessity.
Among the above-mentioned PPOMO processes,the selection of the oxidant plays a crucial role in activity and selectivity.As shown in Fig.1a,the selected oxidants mainly focus on H2O (29.1%),H2O2(16.5%),O2(48.1%).As illustrated in Fig.1b,the selected oxidants were activated to various reactive species for subsequent CH4activation and/or conversion.In PPOMO,CH4was activated mainly by various reactive oxygen species (ROS),such as•OH radicals,O−species,•O2−radicals (Fig.1b).Photocatalytic methane conversion is a redox reaction driven by energetic charge carriers (Fig.1c).The band gaps and band positions of typical semiconductor photocatalysts and relative potentials of various ROS or intermediates were shown [86].
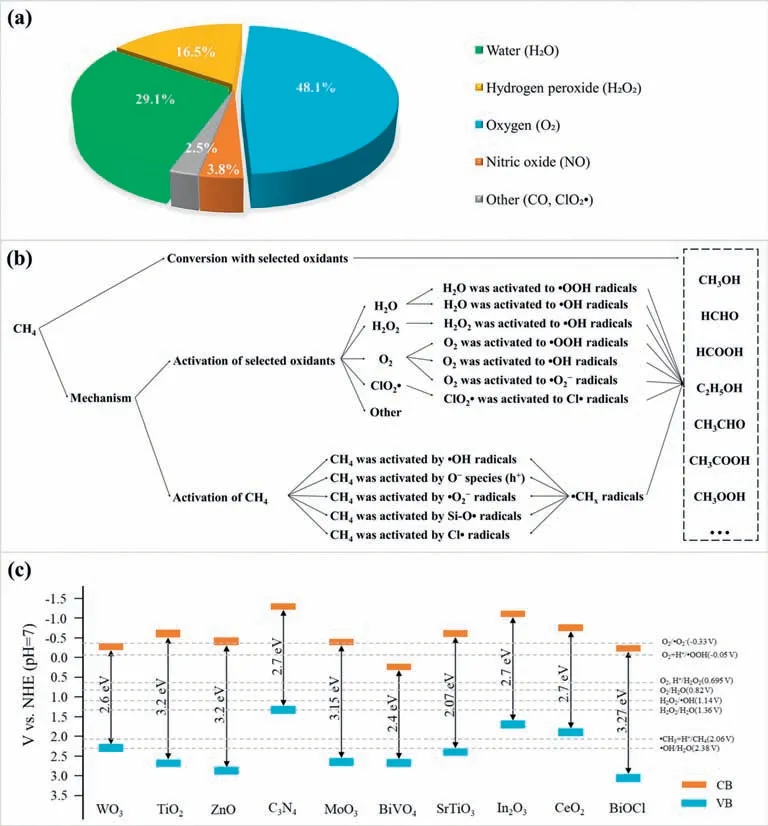
Fig.1. (a) Distribution proportion of different oxidants selected in PPOMO process.(b) Schematic illustration about the activation of selected various oxidants and the activation of methane in PPOMO process.(c) Band gaps and band positions of typical semiconductor photocatalysts and relative redox potentials of photocatalytic methane conversion.Abbreviations: NHE,normal hydrogen electrode;CB,conduction band;VB,valence band.
In this review,we would like to present a comprehensive and systematic summary on the research achievements in photocatalytic partial oxidation of methane to liquid oxygenates from the perspective of the selected oxidants.Meanwhile,the activation of selected oxidants is emphasized and highlighted,and the activation mechanism of methane in PPOMO is summarized.Finally,challenges and perspectives are briefly discussed.We hope this review could provide guidance on how to design the photocatalytic reaction system and how to get a better understanding of reaction mechanism for the development of efficient photocatalytic methane partial oxidation to liquid oxygenates.
2.PPOMO with various oxidants
As mentioned above,the selection of oxidants is important to methane conversion and the overoxidation regulation.In PPOMO process,the most selected oxidants are H2O,H2O2,O2(Fig.1a).According to the oxidant selected,the research progresses of PPOMO are classified into following categories: PPOMO with H2O,H2O2,O2as the oxidant,respectively.Recent progress of PPOMO with various oxidants are summarized in the Table 1.
2.1. PPOMO with H2O as the oxidant
H2O is not only a mild and clean oxidant,but also an effective inhibitor for product overoxidation due to the easier oxygenates desorption [82].Therefore,a number of studies was carried out in exploring H2O as the oxidant for PPOMO.In this PPOMO-H2O system,the most employed semiconductors photocatalysts are WO3,and BiVO4.This section is thus divided into WO3systems,BiVO4systems and other systems based on the photocatalysts used.
2.1.1.WO3systems
WO3was first applied to photocatalytic conversion of methane by Tayloretal.in 1997 [87].The La-doped WO3photocatalyst converted methane to methanol and acetic acid at ∼94 °C and atmospheric pressure under irradiation [88].They highlighted the important role of•OH radicals in both photochemical and photocatalytic processes.In addition,H2O is considered as a supplier of•OH radicals,which is the main species for CH4activation.To gain a deeper understanding of the reaction mechanism,a surface fluorinated WO3was developed and investigated [89].After the fluorination treatment,the CH3OH production decreased (Fig.2a).It was proposed that the surface adsorbed•OH radicals played a dominant role in the PPOMO process (Fig.2b).They highlighted the importance of surface adsorbed•OH radicals for CH4activation and conversion.To clarify the role of La in WO3for the PPOMO,the activity and surface properties of WO3/La were evaluated [90].The CH3OH production of over WO3/La was approximately 2 times higher than that of pure WO3while the CO2production rate was obviously decreased,resulting in a higher CH3OH selectivity (Fig.2c).It was claimed that La doping induced the formation of oxygen vacancies which increased the adsorption of H2O and modification of the basic-acid properties,further enhanced the generation of•OH radicals for CH4activation.To promote the•OH generation,Ag+impregnated WO3was developed.Compared to pure WO3,5%Ag2O@WO3exhibited a higher CH3OH yield.It was proposed that the presence of Ag+on WO3surface enhanced the formation of hydroxyl radicals by suppressing the charge carrier recombination process through formed heterojunction (Fig.2d) [91].The effect of electron scavengers (Fe3+,Cu2+,Ag+) and H2O2species in PPOMO over a mesoporous WO3was studied (Figs.2e-g) [92].The photocatalytic activity of WO3toward CH3OH production could be enhanced 2.5 and 1.7 times by adding Fe3+(1 mmol/L) and Cu2+(0.1 mmol/L),respectively.However,the addition of Ag+had a negative effect on this PPOMO process,which lead to a higher CO2production.
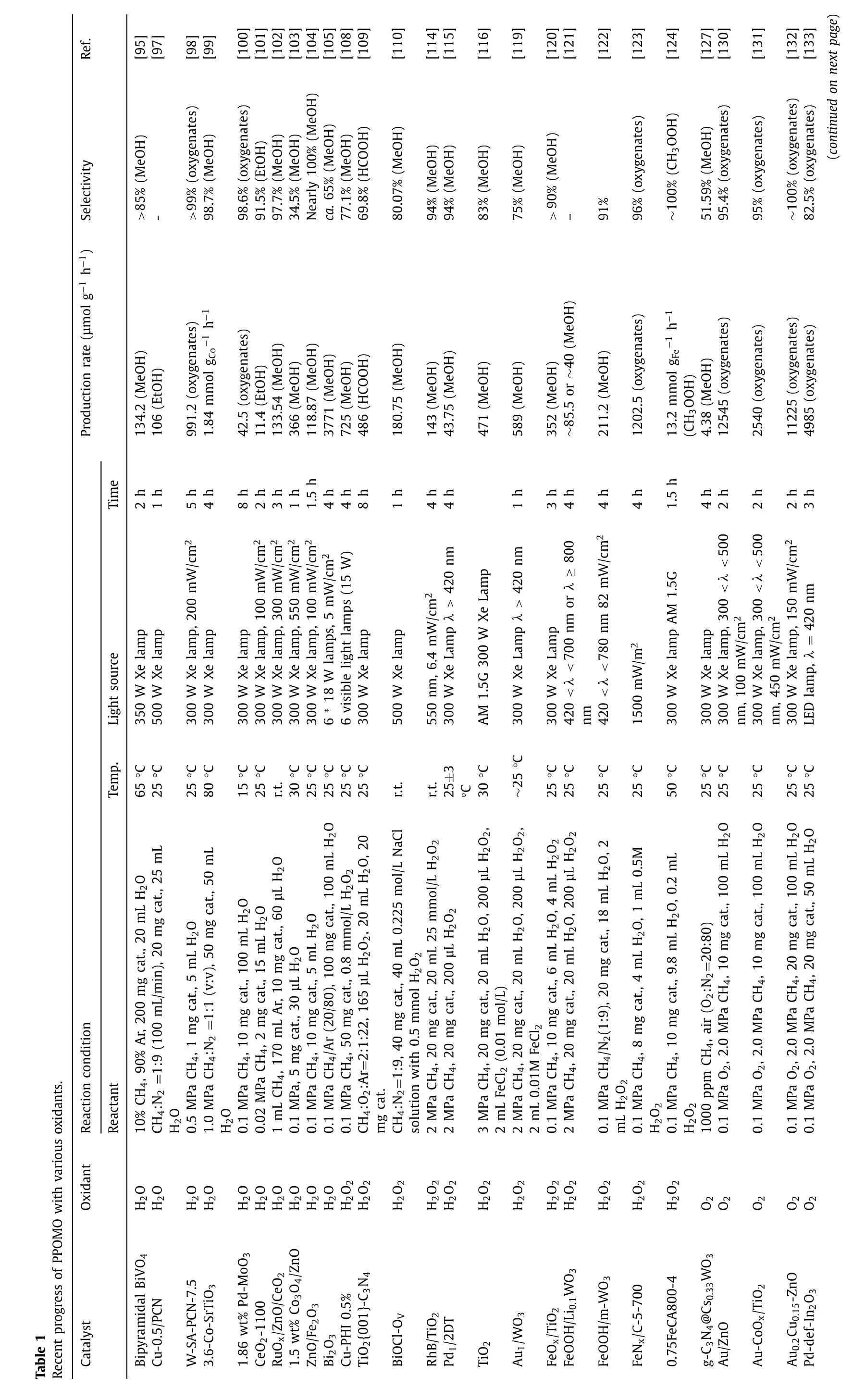
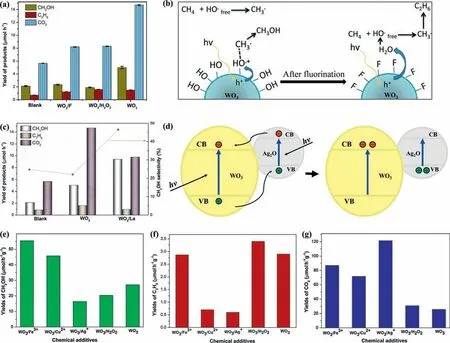
Fig.2. PPOMO with H2O as the oxidant over WO3 systems.(a,b) Yield of products (CH3OH,C2H6,CO2) and proposed reaction pathway for PPOMO on WO3 and WO3/F.Reproduced with permission [89].Copyright 2014,Elsevier B.V.(c) Yield of products and CH3OH selectivity in PPOMO on blank,WO3 and WO3/La.Reproduced with permission [90].Copyright 2016,Elsevier B.V.(d) Proposed mechanism of e−-h+ pair recombination inhibition over Ag2O@WO3 photocatalysts.Reproduced with permission [91].Copyright 2013,Elsevier B.V.(e-g) Yield of products (CH3OH,C2H6,CO2) in WO3/Fe3+,WO3/Cu2+,WO3/Ag+,WO3/H2O2 and WO3 systems.Reproduced with permission [92].Copyright 2014,Elsevier B.V.
2.1.2.BiVO4systems
BiVO4,an alternative photocatalyst to binary oxides as WO3and TiO2,has emerged as a proper candidate for PPOMO.BiVO4was firstly employed to convert methane into liquid oxygenates by Murciaetal.[93].The BiVO4photocatalyst achieved a CH3OH production rate of 21 μmol g−1h−1and a higher selectivity compared to Bi2WO6and Bi2WO6/TiO2-P25 (Fig.3a).A•OH scavenger (nitrite ions,NO2−) was introduced to improve the CH3OH selectivity for PPOMO (Figs.3b and c) [94].In addition,the CH3OH selectivity is even higher than 90% between 60 and 120 min at a relatively high NO2−concentration of 1 mmol/L.It was claimed that the effect of NO2−as a•OH scavenger might limit the overoxidation of produced CH3OH,thereby suppressing the CO2formation and improving the CH3OH selectivity.Crystal facet engineering was also reported to enhance the activity and selectivity of CH3OH during PPOMO process (Fig.3d) [95].Bipyramidal BiVO4comprising {102}and {012} surface facets was more active and selective for CH4to CH3OH compared to the platelet BiVO4with {001} facets as its top and bottom surface.Compared to the other two photocatalysts,the bipyramid possesses more surface adsorption oxygen species.A CH3OH production of 111.9 μmol g−1h−1with a selectivity of 850%was achieved over the bipyramidal BiVO4photocatalyst.It was proposed that the bipyramids combine efficient extraction of holes from the entire surface of the microcrystal and intermediate surface reactivity leading to high•OH production from H2O oxidation and high activity/selectivity for PPOMO.Other Bi-based photocatalysts,such as,Bi2WO6,Bi2WO6/TiO2[93] and Bi/V modified beta zeolite [96],were also developed and reported.The Bi2WO6and Bi2WO6/TiO2samples exhibited good ability to convert methane,while the Bi2WO6,Bi2WO6/TiO2-P25 tend to produce CO2and result in a poor selectivity towards CH3OH.Compared with the pureβ-zeolites (HBEA),Bi-V modified HBEA showed a significant decrease of the undesired CO2production and an improvement of CH3OH selectivity (Fig.3e),which could be attributed to the formed V2O5/BiVO4heterostructure and the depressed nonoxidative coupling of CH4to C2H6.
Fig.3. PPOMO with H2O as the oxidant over BiVO4 systems.(a) Production rates and selectivity with BiWO6,BiWO6-TiO2 and BiVO4.Reproduced with permission [93].Copyright 2014,American Chemistry Society.(b) CH3OH,CO2 and C2H6 production rates obtained during the blank and photocatalytic tests with BiVO4,in the absence and in the presence of with NO2−(1 mmol/L).(c) Selectivity with BiVO4 (A),BiVO4 with 0.5 mmol/L NO2−(B) and BiVO4 with 1 mmol/L NO2−(C).(b,c) Reproduced with permission [94].Copyright 2015,Royal Society of Chemistry.(d) Production rates of CH3OH,CO2 as well as CH3OH selectivity over different structured BiVO4 with bipyramid,thick platelet,and thin platelet morphologies.(e) Production rates of CH3OH,C2H6,and CO2 as well as CH3OH selectivity over HBEA,V-HBEA,and Bi-V-HBEA photocatalysts.(d,e) Reproduced with permission [5].Copyright 2019,Elsevier Inc.
2.1.3.Othersystems
It was reported that an ethanol production rate of 106 μmol g−1h−1was achieved within-situgenerated H2O2on Cu-0.5/PCN photocatalyst (Figs.4a-c) [97].A negligible H2O2production was obtained on Cu-0.5/PCN compared with that of PCN,while a significant increase of ethanol yield was achieved over the Cu-0.5/PCN.The authors claimed that the Cu species not only decomposed thein-situgenerated H2O2to form•OH,but also acted as the active sites for CH4adsorption and activation,which avoided the deep mineralization and further enhanced the photocatalytic anaerobic methane conversion.H2O was also claimed to be activated to•OH radicalsviaH2O2(H2O → H2O2→•OH) for CH4activation on PCN supported W single-atom photocatalyst (W-SAPCN),SrTiO3supported Co particle (Co-SrTiO3) [98,99].The Wδ+was claimed to be active sites for CH4activation and•OH production.Thein-situIR results indicated the benefit of W-SA PCN for CH4adsorption.The interfacial sites between Co particles and SrTiO3are reported forin-situ•OH generation and CH4adsorption,which attributed to enhanced CH3OH production.Compared with the raw SrTiO3,3.6-Co-SrTiO3exhibited superior ability of CH4adsorption and activation according to thein-situDRIFT results.Moreover,according to the experiments they carried out using isotopically labeled reactants (D2O,H218O,or CD4),it was claimed that the O in the hydroxyl group of the generated CH3OH originates from water,whereas H2O and CH4contribute 90% and 10% to the H in the hydroxyl group of the generated CH3OH,respectively.In another system,H2O was reported to be activated to•OOH radicalsviaH2O2(H2O → H2O2→•OOH) for CH4conversion on Pd modified MoO3(Pd-MoO3) when the reaction temperature was 15 °C [100].The proposed mechanism indicated that PdO species on Pd-MoO3played an essential role in the suppression of overoxidation.From the corresponding1H NMR spectrum of the isotope labeling experiments,they claimed that the CH3OH product came from CH4oxidation.The ceria nanoparticles with oxygen vacancies was reported to evoke photocatalytic methane conversion to C2 liquid oxygenates (Fig.4d)[101].Compare with the CeO2-raw,a higher ethanol production was achieved over CeO2-x with oxygen vacancies.They proposed that the oxygen vacancies induced H2O oxidation to•OH(H2O →•OH) and further enhanced the photocatalytic activity.RuOxdecorated ZnO/CeO2nanorods (RuOx/ZnO/CeO2) [102],Co3O4nanoparticles decorated ZnO (Co3O4/ZnO) [103],in-plane Z-scheme hetero-ZnO/Fe2O3[104] and Bi2O3[105] were developed for selectively conversion of CH4to CH3OH.For these systems,the selected H2O oxidant was claimed to be oxidized to•OH radicals(H2O →•OH) for CH4conversion.The RuOx species was claimed to be a beneficial site for•OH generation from H2O oxidation.It is proposed that Co3O4plays as the active sites of stabilization of•CH3adsorption and promotion of CH3OH desorption to suppress overoxidation.The oxygen vacancy in the Bi2O3surface was claimed to be the active site for the adsorption and activation of oxygen.Moreover,the oxygen vacancy could also enhance the adsorption and fixation of CH4.Generally,•OH generation from H2O oxidation is responsible for CH4activation and conversion.Meanwhile,thein-situgenerated H2O2from water oxidation reaction have shown potential for efficient PPOMO.Element doping,crystal plane engineering,cocatalyst modification,and heterojunction construction presents as effective strategies for H2O activation.H2O is generally considered as a mild and clean oxidant for PPOMO,while endeavors are highly needed to enhance activity and sustainability.
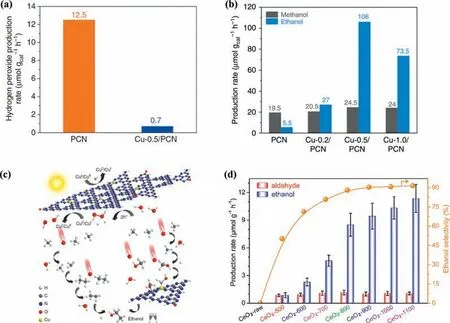
Fig.4. (a) Photocatalytic anaerobic H2O2 production over PCN and Cu-0.5/PCN.(b) Liquid products of CH4 conversion over PCN and Cu-X/PCN.(c) The proposed mechanism for photocatalytic anaerobic CH4 conversion over Cu-0.5/PCN.(a-c) Reproduced with permission [97].Copyright 2019,Springer Nature Limited.(d) Production rates of the photocatalytic products aldehyde (CH3CHO) and ethanol (CH3CH2OH) over CeO2-raw and CeO2-x photocatalysts.Reproduced with permission [101].Copyright 2020,MDPI(Basel,Switzerland).
During the process of PPOMO with H2O as the oxidant,the•OH radical generated from water oxidation plays an important role in PPOMO activity and selectivity.The C-H bond of CH4is activated by•OH radical into•CH3radical which will be further converted to CH3OH (Eqs.1 and 2).Hence,a certain amount or concentration of•OH radical is really needed for CH4activation and conversion.La doped WO3,Ag+impregnated WO3,BiVO4with the expose {001}facet were reported to be useful for charge carrier separation and•OH radical generation.Cu species,Wδ+,interface between Co particles and SrTiO3,RuOxand Ce particle were claimed to be active sites for•OH radical generation.However,the excess•OH radical would cause the overoxidation of formed CH3OH and a low PPOMO selectivity (Eq.3).NO2−was reported as a•OH scavenger for•OH regulation.Co3O4was also reported as an active site for overoxidation suppression.In short,the amount or concentration of•OH radical should be well regulated to a proper level.
2.2. PPOMO with H2O2 as the oxidant
As analyzed and discussed above,•OH radicals are crucial to the activation and conversion of methane.H2O2,as an efficient•OH generator,has been frequently selected for PPOMO [106–110].The effect of H2O2for PPOMO was investigated [91,92,111] and the content or concentration of added H2O2should be regulated to a relatively appropriate level [112].Meanwhile,the efficient utilization of added H2O2should be also highly emphasized.
Sun developed and employed the ZnO nanosheets for PPOMO with H2O2as the oxidant [112].They then investigated the effect of added H2O2.The maximal liquid oxygenates production rate of 2.21 mmol g−1h−1was achieved when adding 500 μL H2O2in this system (Figs.5a and b).However,when more H2O2was added,the products overoxidation to CO2would take place and the selectivity of liquid oxygenates would decrease due to the excessive H2O2.The selected H2O2was activated by photogenerated electrons to•OH and•OOH radicals,which would be reacted with formed•CH3to produce liquid oxygenates.The selected H2O2was also claimed to be activated by photogenerated electrons over a fluffy metal free carbon nitride [113],a dye-sensitized TiO2[114] and an atomicscale Pd supported on 2D titania sheet (Pd1/2DT) [115].Hu synthesized the fluffy mesoporous graphitic carbon nitride (g-CN) and investigated its performance of PPOMO at a mild condition (35 °C)in the presence of H2O2.To estimate the effective utilization ratio of H2O2for CH3OH production,the Gain Factor (GF,the ratio of the molecular amount of CH3OH produced to the molecular amount of H2O2consumed) was introduced.To develop lowenergy input route for PPOMO,Wang developed the dye-sensitized TiO2(RhB/TiO2) and obtained a CH3OH production rate of 143 μmol g−1h−1with 94% selectivity under the irradiation of green light (550 nm).The selected H2O2was claimed to be decomposed to•OH radicals by photogenerated electrons transferred from RhB to the CB of TiO2.The oxygen vacancy in Pd1/2DT was claimed to be not only facilitated the visible light harvesting of 2DT but also stabilized atomic Pd1species.
The efficient utilization of the oxidant of H2O2is related closely to the PPOMO performance.As we known,Fenton reaction could effectively utilize H2O2to form sufficient•OH radicals for water treatment and pollutants removal [116–119].Recently,a synergistic photocatalysis–Fenton reaction for efficient PPOMO under ambient conditions was reported by Wang (Fig.5c) [116].They found that photocatalysis–Fenton reaction contributed to an excellent performance of 471 μmol g−1h−1for selective production of CH3OH from CH4on TiO2compared to Fenton reaction,photo-Fenton reaction,photocatalysis reaction.The introduction of Fe2+to this reaction system promotes the activation of H2O2to•OH radicals for PPOMO.And the optimal ratio of Fenton reagents H2O2/Fe2+was determined to be 20:1.The excess H2O2would react with•OH to produce•O2−and further give rise to the overoxidation of CH3OH.
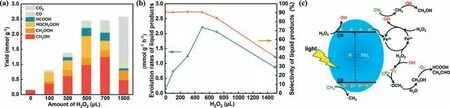
Fig.5. PPOMO with H2O2 as the oxidant.(a,b) Performance and proposed mechanism of PPOMO over ZnO nanosheets.Reproduced with permission [112].Copyright 2021,American Chemical Society.(c) Proposed mechanism of photocatalysis-Fenton reaction for selective conversion of CH4 to CH3OH over TiO2.Reproduced with permission[116].Copyright 2020,Royal Society of Chemistry.
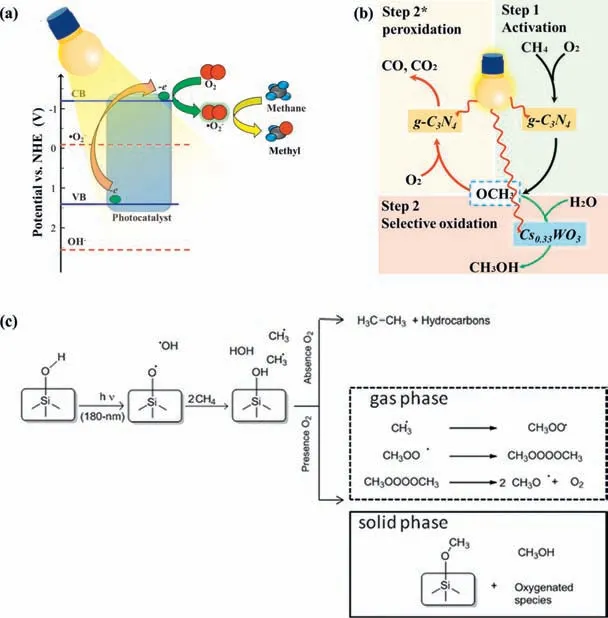
Fig.6. PPOMO with O2 as the oxidant in gas phase systems.Proposed activation mechanism of CH4 (a) and reaction scheme (b) over g-C3N4-decorated Cs0.33WO3.(a,b)Reproduced with permission [127].Copyright 2019,American Chemical Society.(c) Proposed mechanism for deep UV conversion of CH4 on silica surface.Reproduced with permission [128].Copyright 2011,American Chemical Society.
Besides,a series of Fe species modified catalysts were reported to efficiently utilize H2O2for methane activation and conversion[120–124].Tangetal.developed the metal oxides anchored on TiO2as photocatalysts for PPOMO with H2O2as the oxidant [120].In this system,the optimal ratio of CH4:H2O2was determined to be 8.75:1.The excess H2O2would produce more CO2,which would lead to a low selectivity to CH3OH.The Fe species not only promoted the activation of H2O2,but also enhanced charge transfer and separation,which further improved the CH3OH selectivity.H2O2was activated by photogenerated electrons on FeOxand form•OH radicals for PPOMO.Visible-light responsive WO3was also developed and applied to PPOMO with H2O2as the oxidant [121,122].They claimed that FeOOH could enhance the activation of H2O2to form•OH radicals for CH4activation.H2O2is well known for its homolytic cleavage into•OH,but this reaction always accompanied by unexpected O2generation.To make full use of the•OH generated from H2O2,a heterogeneous Fenton-type Fe-based catalyst containing Fe-Nxsites and Fe/Fe3C nanoparticles was fabricated and studied in PPOMO [123].They claimed that the Fe-Nxin the low spin state provides active sites for H2O2activation and•OH generation.The photocatalyst was also claimed to be effective for CH4adsorption.Xie developed the Fe clusters anchored on carbon aerogel (0.75FeCA800-4) for PPOMO with a near 100%CH3OOH selectivity [124].The positively charged Fe clusters could not only promote the activation of H2O2to•OH,but also donate electrons for CH4activation.The three-dimensional carbon aerogel was claimed to be favorable to the adsorption of CH4and release the products.On the Cu-PHI photocatalyst,the Cu atom was reported to be active sites for the formation of hydroxyl radicals attributed to the adsorption of hydroxyl group.
H2O2,as an efficient•OH radical supplier,has been well selected and applied to CH4activation and conversion.The efficient utilization of H2O2in PPOMO attracts increasing attention whether activated by photogenerated electrons or combined with Fentonlike reaction.On one hand,the efficient activation of selected H2O2could bring sufficient•OH radical should be regulated at a proper level of concentration or content.The concentration of added H2O2should be optimized.Single-atom Pd,various Fe species were introduced to be electrons acceptors for efficient H2O2activation and sufficient•OH radicals acquisition.On the other hand,the amount or concentration of formed•OH radicals should be well regulated at a proper level due to the overoxidation.The loading amount of Fe species or electrons promoter was optimized to regulate the concentration of•OH radicals.These progresses and the discussion of H2O2utilization above might provide instructive insights for further study about PPOMO with H2O2as the oxidant.
2.3. PPOMO with O2 as the oxidant
O2is usually employed as the oxidant in varieties of oxidation reactions because of the ability to form reactive oxygen species(ROS) [125].From the availability,operating safety and the ability to form ROS,O2is an attractive and ideal oxidant for efficient PPOMO.As shown in Fig.1a,O2as the oxidant accounts for 48.1%of the published PPOMO articles.It is therefore important to figure out the role played by O2,and more attention should be paid to the activation of O2in PPOMO process.According to the different reaction systems,the related research works could be classified into two categories: Gas phase systems and liquid phase systems.
2.3.1.Gasphasesystems
In gas phase systems,methane was reported to be oxidized to CH3OH [60,126–129],HCHO [61–67],HCOO [68] with O2as the oxidantviaphotocatalysis.
CH4conversiontoCH3OH: Brazdil firstly achieved the optimal CH3OH conversion rate of 6 μmol/h on CuMoO4in the presence of O2at 100 °C under UV irradiation [60].Mo-containing porous TiO2exhibited a higher CH3OH production than pure TiO2in the presence of O2UV irradiation,which could be attributed to the broadened light absorption [126].Recently,Wuetal.reported that g-C3N4decorated Cs0.33WO3(g-C3N4@Cs0.33WO3) selectively converted low concentration CH4into CH3OH [127].The selected oxidant of O2was activated to•O2−which dominated the C-H bond cleavage and promoted the generation of CH3O•intermediate.More photogenerated electrons from Cs0.33WO3protected CH3O•from overoxidation and selectively enhanced the conversion ratio from CH3O•into CH3OH (Figs.6a and b).
A series of zeolites were also developed and applied to PPOMO with O2as the oxidant in gas phase systems [128,129].It was reported that a deep UV photolysis of CH4into C1 liquid oxygenates over beta zeolites containing internal silanol groups,and the product selectivity toward C1 liquid oxygenates (CH3OH,HCHO,HCOOH) was over 95% with the addition of O2[128].Beta-,delaminated ITQ2 and ITQ6,medium-pore ZSM5 zeolites,mesoporous MCM41 silicates,non-porous TiO2were employed as the catalysts for converting CH4into C1 liquid oxygenates [129].The UV irradiation induced the homolytic cleavage of surface hydroxyl group in the micropores of beta zeolite,leading to the formation of silyloxyl radicals (≡Si-O•) that will generate•CH3from CH4(Fig 6c).It was claimed that confinement and porosity increased the proportion of C1 liquid oxygenates adsorbed onto the solid and reduced the contribution of the gas-phase products.And the presence of O2in the systems considerably increased the selectivity toward C1 liquid oxygenates and significantly reduced the production of ethane and other alkanes.
CH4conversiontoHCHO: PPOMO (HCHO) with O2as the oxidant in gas phase systems over various catalysts were summarized in Table 2.In the 1990s,Wada developed a series of metal oxides for PPOMO (HCHO) with O2as the oxidant in gas phase [61–66].To investigate the correlation between the activity and the surface state,the effects of the preparation method (ED: evaporation to dryness;IW: incipient wetness;SG: sol-gel) was studied.They claimed that only four-coordinated vanadium oxide surface species rather than crystalline V2O5act as the catalytic active sites for PPOMO.A vanadium-supported SBA-15 was developed and investigated for the PPOMO by Martinez [67].The isolated four-coordinated V5+species was claimed to be the active centers.
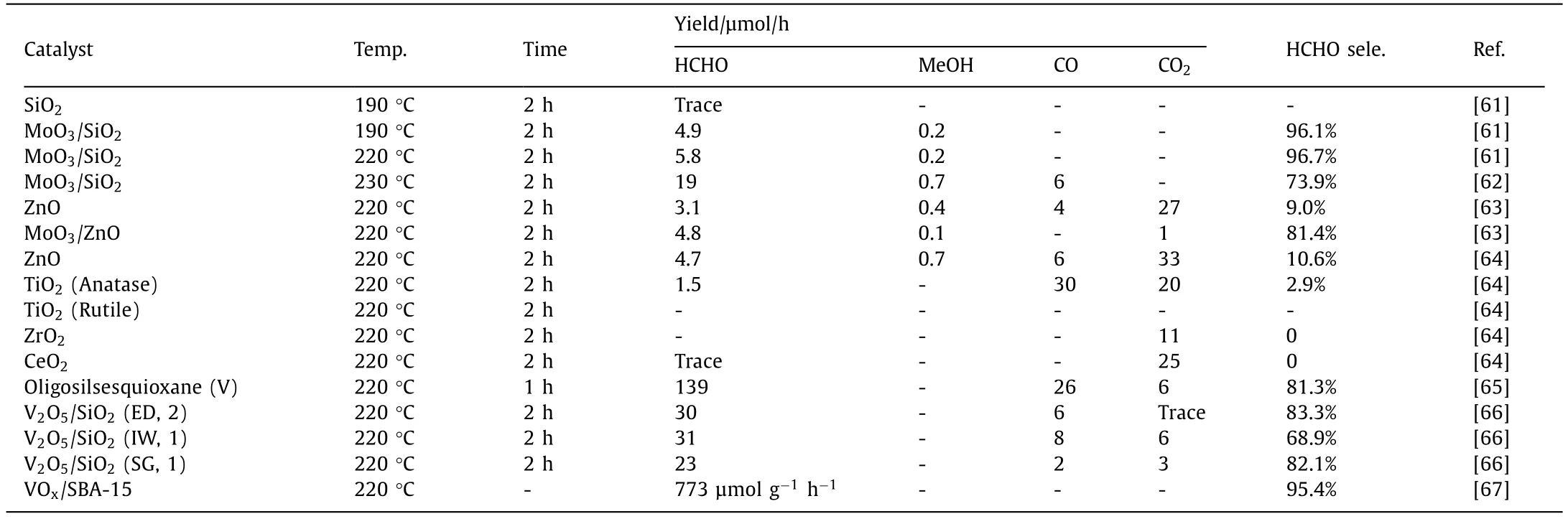
Table 2 PPOMO (HCHO) with O2 as the oxidant in gas phase systems over various catalysts.
During the PPOMO processes with O2as the oxidant in gas phase,the photothermal effect should not be ignored.More attention should be paid to how the photothermal effect is involved in the activation of oxygen molecules and how to promote the conversion of methane or improve the selectivity of products.
2.3.2.Liquidphasesystems
In liquid phase systems,produced liquid oxygenates are easy to be removed from catalyst surface by the solvent H2O,which would inhibit the products overoxidation due to the easier oxygenates desorption.The selected O2was activated to various ROS,•OOH radicals [130–136],•OH radicals [137–143],and•O2−radicals[144–155].
O2wasactivatedto•OOHradicals: Ye reported that photocatalytic CH4conversion to liquid oxygenates in H2O with a productivity of ∼12500 and 2540 μmol g−1h−1over Au/ZnO and Au-CoOx-TiO2,respectively [130,131].They claimed that molecular O2rather than H2O,is the source of oxygen for direct CH4oxidation through the experiments with isotopically labeled O2and H2O.Moreover,they proposed that O2was activated to milder oxidative•OOH radicals compared to•OH radicals which was claimed to be the culprit of products overoxidation (Fig.7a).The generated•OOH radicals will be coupled with•CH3to generate CH3OOH and beneficial to avoid the overoxidation.The CoOxspecies was introduced as a cocatalyst to lower the potential of photogenerated holes to suppress the formation of•OH radicals from H2O oxidation.The selected O2was also reported to be activated to•OOH radicals on binary Au-Cu reactive sites decorated ZnO (Au0.2Cu0.15-ZnO) [132],defective In2O3supported atomic Pd (Pd-def-In2O3) [133],defective TiO2supported atomic Pd (Pd-def-TiO2) [134].They claimed that Cu species serve as photoinduced electron mediators to promote O2activation to•OOH,and Au is a hole acceptor to enhance H2O oxidation to•OH through thein-situEPR and XPS measurements.On the Pd-def-In2O3and Pd-def-TiO2photocatalysts,the oxygen vacancy was reported to be the electron acceptor activating O2to•OOH.
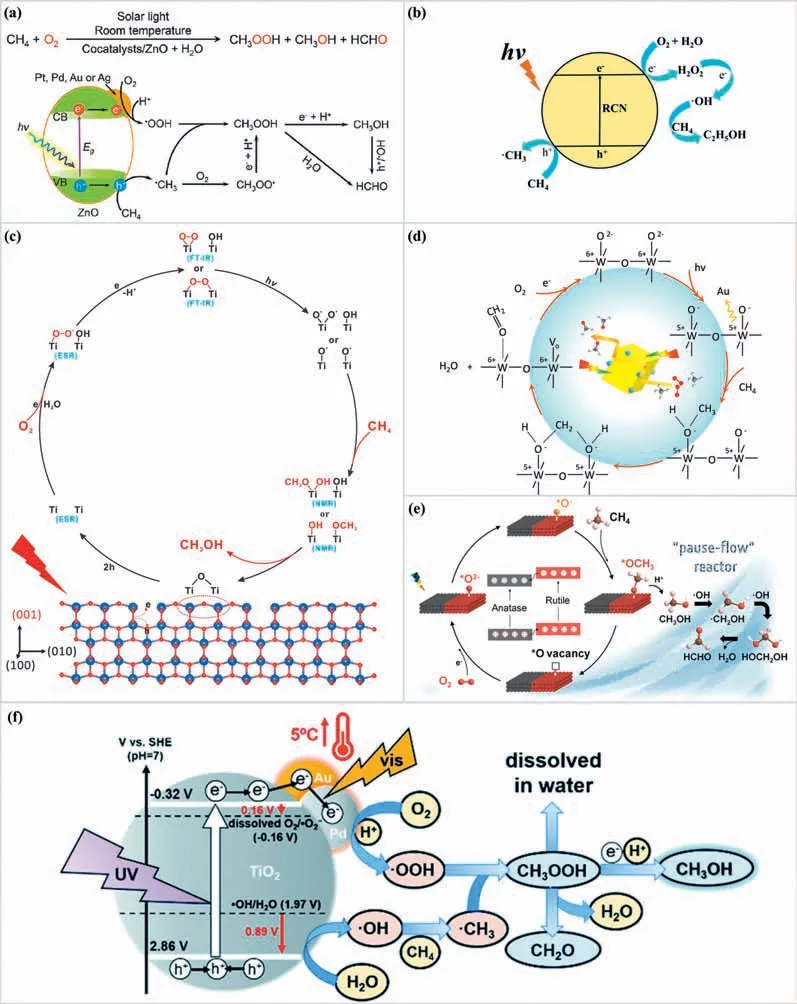
Fig.7. Proposed mechanisms of PPOMO with O2 as the oxidant in liquid phase systems.(a) O2 was activated to •OOH radicals over Au/ZnO.Reproduced with permission[130].Copyright 2019,American Chemical Society.(b) O2 was activated to •OH radicals via H2O2 over nitrogen vacancy-rich carbon nitride.Reproduced with permission [140],Copyright 2021,Royal Society of Chemistry.(c) Proposed photocatalytic mechanism for CH4 oxidation by O2 on the {001} facets of TiO2.Reproduced with permission [143].Copyright 2021,Springer Nature Limited.(d) Proposed reaction mechanism for HCHO production over c-WO3.Reproduced with permission [145].Copyright 2020,Elsevier B.V.(e) Proposed mechanism of photocatalytic CH4 oxidation on A/R-TiO2.Reproduced with permission [147].Copyright 2022,American Chemical Society.(f) Reaction mechanism of photocatalytic CH4 conversion over Au–Pd/TiO2.Reproduced with permission [148].Copyright 2021,Royal Society of Chemistry.
O2wasactivatedto•OHradicals: Au nanoparticles modified ZnO photocatalysts reported by Tangetal.exhibited good performance with CH3OH [137].The selected O2was claimed to be activated into•OH radicals which will attack the CH4molecules or couple with the formed•CH3to CH3OH.They claimed that the O-source of CH3OH was mainly from H2O oxidation rather than molecule oxygen.The loading amount of Au nanoparticles was reported to be optimized to regulate the concentration of•OH radicals and alleviate the product overoxidation.They also developed a quantumsized bismuth vanadate (q-BiVO4) for PPOMO [138],which offered a selectivity of 96.6% for CH3OH or 86.7% for HCHO under optimal conditions.In this system,O2was activated to•OH radicals for CH4activation.Recently,we reported that the effi-cient conversion of methane with H2O2in-situgenerated from oxygen reduction reaction.A record ethanol production rate of 281.6 μmol g−1h−1was achieved over nitrogen vacancy modified carbon nitride (RCN-5) at room temperature [140].Through the investigation of H2O2and•OH intermediates,we proposed that O2was activated to•OH radicalsviaH2O2(O2→H2O2→•OH) (Fig.7b).Moreover,the optimal concentration ofin-situgenerated H2O2was estimated to be 217.1 μmol/L.Furthermore,an excellent ethanol production was achieved over optimized P-doped g-C3N4(CNP-50)under atmospheric pressure and room temperature [141].The Pdoping enhanced the•OH production from O2reduction reactionviaH2O2(O2→H2O2→•OH) and further promoted the ethanol production.A sulfone-modified conjugated organic polymers (SCTTP) was reported to be able to drive O2→H2O2→•OH for CH4activation and conversion to CH3OH and HCOOH [142].A monoiron hydroxyl site immobilized within a metal-organic framework,PMOF-RuFe(OH) was reported to be efficient for CH4conversion to CH3OH with a time yield of 8.81 ± 0.34 mmol g−1h−1and 100% selectivity [143].Compared to PMOF-Ru,PMOF-RuFe(OH) has a hysteretic desorption,suggesting that the mono-FeⅢspecies acts as a binding site for CH4.Besides,the produced CH3OH could desorb rapidly from the MOF matrix in the presence of water.The selected O2oxidant was claimed to be photo-reduced to•OH radicalsviain-situgenerated H2O2(O2→H2O2→•OH).•OH radicals could be originated from both water oxidation reaction and oxygen reduction reaction.Noble metal modification,vacancy introduction,and the quantum size effect have been reported to be effective ways to enhance the•OH radicals acquisition.The nitrogen vacancy modified carbon nitride and P-doped carbon nitride exhibited excellent ability to obtain•OH radicalsviain-situgenerated H2O2for CH4activation and conversion.The regulation of the amount ofinsitugenerated H2O2or•OH radicals could be achieved through optimizing the concentration of introduced nitrogen vacancy and the P doping amount.
O2wasactivatedto•O2−radicals: Ye developed the Agdecorated facet-dominated TiO2(Ag/TiO2{001}) to control the overoxidation of PPOMO [143].Through the operando FT-IR,insituESR,and NMR characterizations,they found that the oxygen vacancies generated on {001} facets could avoid the formation of•CH3and•OH radicals in the following reaction steps (Ti-O2•→Ti-OO-Ti and Ti-(OO) →Ti-O•pairs) to reduce overoxidation (Fig.7c).The selected O2was activated to•O2−radicals which will be stabilized on a metallic cationic site by the oxygen vacancy.A similar speculative mechanism was proposed on the cuboid shaped WO3(c-WO3) by Fan (Fig.7d) [145].O2was activated to•O2−radicals which will be stabilized on the W5+sites for further CH4activation and conversion.Based on the XPS results and band structure,the c-WO3was claimed to have a high O2adsorption capacity.Furthermore,O2could renovate the dissipative crystal-O atom in the continuous reaction within 24 h [146].From thein-situDRIFTS,a higher signal intensity ascribed to the adsorbed formate species was observed on TiO2compared to that of the W-TiO2.That means the generated oxygenates desorb from the W-TiO2easier and a higher oxygenates selectivity.An HCHO production of 8.03 mmol g−1h−1was achieved over a bi-phase catalyst with anatase (90%) and rutile (10%) TiO2in a “pause-flow” reactor [147].The selected O2oxidant was activated to•O2−radicals which will be further activated to O−species for CH4activation (Fig.7e).Through thein-situDRIFTS results and XPS spectra,it was claimed that photothermal synergy with a CH3OH yield of 12.6 mmol g−1h−1was reported on Au-Pd/TiO2[148].Based on the comprehensive study of each component,the reaction mechanism was proposed in Fig.7f.The selected O2was activated to•O2−radicals by photo-excited electrons,then transformed into•OOH radicals which will be coupled with formed•CH3radicals to CH3OOH(O2→•O2−→•OOH).It was claimed that Au–Pd nanoparticles not only help to transfer photo-generated electrons,but also absorbed visible light to increase the catalyst temperature by 5 °C.The temperature rise enhanced the driving force for H2O oxidation into•OH,O2reduction into•O2−,and the reduction of CH3OOH into CH3OH.It was also reported that O2was activated to•OOH radicalsvia•O2−radicals (O2→•O2−→•OOH) on polyoxometalates-based photocatalysts (H4SiMo12O40/TiO2[149],CePMo12O40/TiO2[150]),W-TiO2[151],defective TiO2[152],Cu and W codoped TiO2(Cu-WTiO2) [153],and Pt/WO3[154] during the PPOMO process.Besides,the selected O2was reported to be activated to•O2−radicals at the adjacent oxygen vacancy site on 2D BiOCl photocatalyst based on the density functional theory calculations,DMPO-•O2−ESR test and time-dependent absorption spectra of nitro blue tetrazolium chloride (NBT) oxidation experiments [155].The oxygen vacancy was also claimed to be effective sites for CH4and O2adsorption.
O2could be activated to•OOH radicals,•OH radicals and•O2−radicals for CH4conversion,and the activation mechanism was studied by various characterizations,such as EPR,FT-IR,NMR as well as isotope labeling experiments.Although much understanding about the activation mechanism of the selected O2has beenachieved,more efforts should be made to get a deeper insight into how the O2involves the PPOMO and reaction pathway of the O2.
O2selected as the oxidant for photocatalytic partial oxidation of methane into liquid oxygenates shows superiority.We therefore would like to present some typical cases on the photocatalyst design in PPOMO with O2as the oxidant in liquid phase.The most employed photocatalysts were TiO2,g-C3N4,ZnO,WO3.Co-catalyst decoration,vacancy and/or single-atom introduction,crystal facet engineering,crystal phase engineering,and doping have been demonstrated to be useful for PPOMO.CoOxspecies was adopted to tune the oxidative site of TiO2for suppressing the overoxidation of generated CH3OH [131].Nitrogen vacancy was introduced into g-C3N4to promotedin-situH2O2generation for efficient PPOMO [140].Pd single-atom was used for C-H bond cleavage.The biphase catalyst with anatase (90%) and rutile (10%)TiO2was reported to be useful for the formation of intermediate methanol species [147].The photocatalyst design mainly focuses on the activation of CH4,the activation of selected O2,the intermediates adsorption/desorption and the suppression of products overoxidation.
A comparison of PPOMO with H2O,H2O2,O2is listed in Table 3,the advantages and disadvantages of different oxidants are shown.

Table 3 A comparison of PPOMO with different oxidants.
2.4. PPOMO with other oxidants
Besides the oxidants of O2,H2O,and H2O2,there were some other oxidants (NO [156–158],ClO2•[159] and CO [160]) were also employed for PPOMO.Hu reported that the introduction of NO greatly improved the selectivity of CH3OH production over the VMCM-41 (acid) photocatalyst [156–158].It was proposed that the formed•CH3could easily interact with NO to generate CH3OH on the acid V-MCM-41.Ohkubo developed a two-phase system comprising perfluorohexane and water for PPOMO with a CH4conversion of 99% [159].The yields of CH3OH and HCOOH were 14% and 85%,respectively.Moreover,there were no further COxproducts.Skupinskietal.[160] selected CO as the oxidant and reported the direct CH4photocarbonylation experiments which gave CH3CHO in benzene under UV irradiation over rhodium RhCl(CO)(PR3)2complexes (R=alkyl,Ph,or OPh).
3.Activation mechanisms of CH4
As we know,the CH4molecules is difficult to activate,convert and utilize.It is therefore important to make out the activation mechanism of CH4.Up to now,CH4was reported to be activated mainly by•OH radicals,O−species (photogenerated holes),•O2−radicals and other species.
3.1. CH4 was activated by •OH radicals
It is generally acknowledged that•OH radicals can abstract the H atom from CH4.The reactive•OH radicals were mainly generated from H2O oxidation,H2O2decomposition,O2reduction.
When H2O was selected as the oxidant,CH4can be mainly activated by•OH radicals generated from H2O oxidation.Taylor firstly reported the activation mechanism of CH4and preliminary verified by adding the•OH generator of H2O2[87].Similar mechanism of CH4activation was proposed on WO3-based photocatalysts [87–92,111],BiVO4-based photocatalysts [93–96],CeO2[101],Co3O4/ZnO [103],and Bi2O3photocatalyst [105].A slight difference was found on the activation mechanism of CH4over Cu/PCN and W-SA-PCN,CH4molecules was claimed to be activated by•OH radicals from H2O oxidationviaH2O2(Fig.5c) [97,98].
When H2O2was selected as the oxidant,CH4can be mainly activated by•OH radicals generated from H2O2activation.H2O2could decompose by UV-irradiation or photogenerated electrons to•OH radicals for CH4activation (Figs.6d and e).Focusing on the efficient utilization of H2O2and generation of abundant•OH radicals,various reaction systems and photocatalysts were developed.Especially,a series of Fe species decorated photocatalysts (FeOx/TiO2[118],FeOOH/Li0.1WO3[119],FeOOH/WO3[120],FeNx/C-R-T [121] and 0.75FeCA800-4 [122]) were developed for coupling with Fenton reaction to obtain an efficient utilization of H2O2.Meanwhile,it is worth noting that the concentration of H2O2should be regulated to a proper level to avoid the products overoxidation.
When O2was selected as the oxidant in liquid phase (CH4-O2-H2O),CH4can be mainly activated by•OH radicals generated from H2O oxidation or O2reduction.CH4was reported to be activated by•OH radicals from O2reduction over AuCu-ZnO [132],Pd-def-In2O3[133] based on thein-situEPR investigation.CH4was reported to be activated by•OH radicals from O2reduction over q-BiVO4[138],Au/BP [139].In addition,it was claimed that CH4was activated by•OH radicals generated from both H2O oxidation and O2reduction on Aux/ZnO [137].According to the study on intermediates ofin-situgenerated H2O2and•OH radicals,we proposed that CH4molecules was activated by•OH radicals from O2reductionviaH2O2over nitrogen vacancy-rich graphite carbon nitride[140,141].
3.2. CH4 was activated by O−species (photogenerated holes)
It was first reported that CH4was activated by the hole center of the O−species underγirradiation during the PPOMO process [59].The active O−species can be formed by trapping photogenerated hole with lattice oxygen [64].CH4was reported to be activated by O−species on some transition metal oxides,MoO3[61,62],vanadium oxide [66,67,156–158],ZnO [63,112,130],TiO2[131,143],WO3[145].The O−species dominated activation mechanism of CH4is shown in Eq.4 and Fig.8a.The O−species could abstract a H atom from CH4to form•CH3,which would take part in the subsequent reaction to liquid oxygenates.
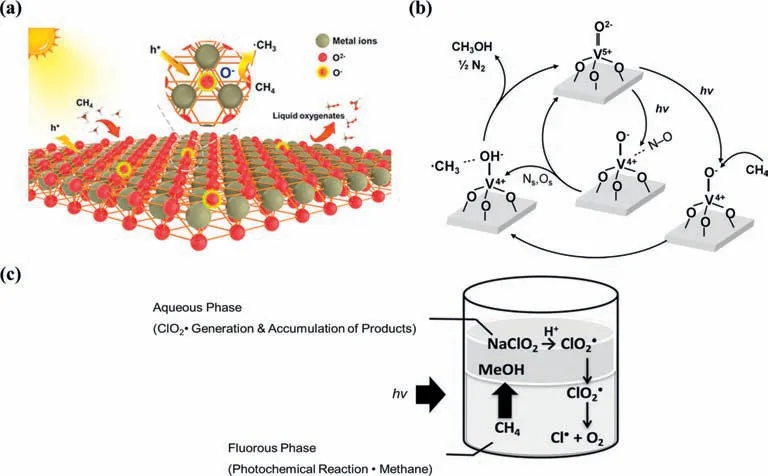
Fig.8. (a) CH4 can be activated by O−species on oxide semiconductors during the PPOMO process.Reproduced with permission [112].Copyright 2021,American Chemical Society.(b) Proposed reaction mechanism for the PPOMO with NO on V-MCM-41(acid).Reproduced with permission [158].Copyright 2013,Elsevier B.V.(c) PPOMO with ClO2•as the oxidant in the two-phase system comprising perfluorohexane and water.Reproduced with permission [159].Copyright 2018,Wiley-VCH.
The CH4activation was reported to be activated by tetrahedrally coordinated vanadium oxide species (O−species) over vanadiumcontaining MCM-41 [158].It was claimed that V4+species and•CH3formed by UV irradiation of the exposed VO4species with methane interact easily with NO,resulting in the selective formation of CH3OH.The charge transfer excited triplet state (V4+-O−)∗species,especially the O−hole trap center reacts with CH4,leading to a break in the C-H bond of CH4to form V4+-OH and•CH3radicals (Fig.8b).The∗OH species then released from V4+-OH would react with the•CH3radicals to form CH3OH.V2O5/SiO2[66],mesoporous VOx/SBA-15 [67],V-MCM-41 mesoporous molecular sieves[156,157] were also reported to be active for CH4activation with the tetrahedrally coordinated vanadium oxide species (O−species).
Ye claimed that CH4was activated by O−species (photogenerated hole centers) of ZnO rather than•OH radicals through a comparison experiment and DFT calculations [130].The comparison experiment of•OH radicals generation using coumarin as probe molecule revealed that cocatalyst/TiO2were more efficient than cocatalyst/ZnO,which was inconsistent with the photocatalytic activities.And the DFT calculations showed that the reaction energy barrier of H abstraction of CH4by O atoms on ZnO is lower than that of TiO2,which was in accordance with experimental results of photocatalytic CH4oxidation.It was therefore claimed that CH4activation over ZnO was dominated by O−species.Specially,twodimensional oxide semiconductors (ZnO nanosheets) are designed to obtain abundant active O−species for activating C−H bond of CH4[112].Thein-situEPR study verified that lattice O atoms could capture photoexcited holes and generate active O−species,which will abstract H from CH4to form•CH3radicals.
It was reported that CH4was activated by photogenerated holes(O−species) from the VB of TiO2to•CH3radicals,which would react with formed ROS to produce liquid oxygenates [116,118,149–153].On the Au-CoOx/TiO2,it was claimed that the photogenerated holes on TiO2rather than Co3O4could oxidize CH4to•CH3according to DFT calculations.More specifically,on TiO2{001}samples,Ti-O••O-Ti species were claimed to activate CH4molecule through the ESR and NMR measurement Fig.7c) [143].Under UV irradiation,the surface oxygen (Ti-O-Ti) of TiO2{001}sample was oxidized by photoinduced holes to form oxygen vacancies (Eq.5),which can stabilize the formed•O2−(Eq.6).And the Ti-O••O-Ti was considered as the active species for CH4activation.Details are as following Eqs.7–9.While on TiO2(101)samples,Ti-O•species were claimed to activate the CH4following the Eqs.10 and 11.The formed•CH3and•OH were claimed to be unfavorable to liquid oxygenates selectivity.
It was also reported that the CH4was activated by O−species on Au/c-WO3according to the isotope characterization,XPS spectra andin-situESR test (Fig.7d) [145].On the crystal-O exposure site of c-WO3,surface W5+(electron) and O−ion (hole) centers were generated through the charge separation under irradiation.Then the formed O−ion (hole) would engage in H-atom abstraction from CH4by being inserted into the C-H bond.The formed surface methoxy (-O-CH3) would take part in the subsequent reaction for HCHO formation.Moreover,the long-time (24 h) stability for HCHO yield was explained to the timely renovation of dissipative crystal-O atom by O2.The cocatalyst of Au was claimed to promote high O−ion (hole) formation through thein-situESR.On the FeOOH/Li0.1WO3[119] or FeOOH/m-WO3[120],CH4was claimed to be activated to•CH3radicals by both photogenerated holes (O−species) and•OH radicals.The main product of CH3OH was generated through the reaction between formed•CH3radicals and•OH radicals.
3.3. CH4 was activated by other species
It was reported that CH4was activated by the•O2−radicals on g-C3N4-decorated Cs0.33WO3[127] and oxygen vacancy modified BiOCl [155] under irradiation during the PPOMO process with O2as the oxidant.The selected oxidant of O2was activated to•O2−radicals,which would subsequently activate CH4to form CH3OH.The difference is the reaction pathway towards CH3OH.On the g-C3N4@Cs0.33WO3photocatalyst,it was claimed that CH4was activated to methoxyl radicals,which would be converted to CH3OH Fig.6a).On the BiOCl-OV photocatalyst,it was claimed that•O2−radicals attacked CH4and initiated the breaking the CH3-H and the O-O bond together.CH4was activated to adsorbed CH3OH,which would desorb from the catalyst surface easily (Eqs.12 and 13).However,more direct evidence should be provided for a further confirmation of this•O2−radical dominated CH4activation mechanism.
It was reported that CH4could be activated to•CH3by silyloxyl radicals (≡Si-O•) on series of zeolites [128,129].Under deep UV irradiation (λ<200 nm),the surface hydroxy group (≡Si-OH) was broken into ≡Si-O•radicals,which could activate CH4to•CH3by H abstraction.The formed ≡Si-O•radicals and•CH3radicals then combined and produced CH3OH (Fig.6c and Eqs.14–17).
The ClO2•was reported to be an efficient oxidant for PPOMO with a 99% conversion [159].The methane oxygenation is initiated by the photochemical Cl-O bond cleavage of ClO2•to generate Cl•and O2.The produced Cl•reacted with CH4to form a•CH3,CH3OH and HCOOH were given by the radical chain reaction (Fig.8c).The fluorous solvent plays an important role of inhibiting the deactivation of reactive radical species such as Cl•and•CH3.
4.Conclusions and prospects
Photocatalytic partial oxidation of methane to liquid oxygenates(CH3OH,C2H5OH,HCHO,CH3CHO,HCOOH,CH3OOH,etc.) is a sustainable and potential technology towards addressing the increasingly sever energy shortage and environmental deterioration.In this review,we have highlighted research progresses in the PPOMO from the perspective of selected oxidants (including H2O,H2O2,O2,NO,ClO2•and CO).Moreover,the activation of selected oxidant (such as,H2O2and O2) is also underlined to gain a deeper understanding on reaction mechanism.We then discussed the activation mechanism of CH4,which was mainly rely on•OH radicals and active O−species.
Although remarkable achievements have been made in PPOMO,there are still many challenges to be addressed in exploring effi-cient reaction systems to its full potential.To make a small step forward and broaden the research scope of the PPOMO,our discussions are as follows:
(1) The reaction systems should be well designed.Temperature,pressure,light source,reaction time as well as the selection of oxidant take an important part in efficient PPOMO.For example,in the CH4-O2-H2O systems,a pressurized reactor was employed to increase the solubility of CH4and O2in H2O.The amount of H2O is significant to the desorption of generated products (CH3OH,C2H5OH,CH3OOH,etc.)to avoid the overoxidation.Reaction time is also an indispensable factor for the activity and selectivity of PPOMO.A“pause-flow” reactor was also reported for efficient CH4conversion.Hence,elaborate design of the reaction systems and precise regulation of each component are exactly needed for achieving an efficient PPOMO with high activity and high selectivity.
(2) The design of novel photocatalysts is in urgent demand.ZnO nanosheets was designed to generate abundant active O−species for activating C-H bond of CH4.A series of containing Fe species photocatalysts were developed for effi-cient H2O2utilization to enhance the PPOMO performance.Ag-decorated facet-dominated TiO2was reported to efficient and selective photocatalytic CH4conversion to CH3OH with O2by controlling overoxidation.Taking the adsorption of substrates into account,the design of photocatalyst surface should be highly heeded.TiO2,WO3,ZnO are emerging as efficient metal oxide photocatalysts for CH4activation in PPOMO.The acquisition and regulation of the reactive oxygen species could be addressed mainly through the well design of photocatalyst and reaction systems.Crystal facet engineering,Fe or Cu species,single-atom catalyst,vacancy,defects,and three-dimensional structure have been demonstrated to be effective methods to construct active sites for CH4adsorption and O2adsorption.The solvent may tune the hydrophilic/hydrophobic properties of the photocatalyst surface and the adsorption of intermediate species.Moreover,the metal sites immobilized with MOF was also reported to be efficient for PPOMO.Meanwhile,given the sunlight spectrum distribution and long-term sustainable reaction,visible light-responsive and stable photocatalyst should be developed.
(3) More attention is deserved should be paid on the PPOMO with O2as the oxidant in liquid phase due to lower energy input and higher conversion &selectivity.Photocatalytic conversion of methane with H2O2in-situgenerated(•OH radicals) from whether oxygen reduction reaction or water oxidation reaction has shown great potential in this field.Sufficient•OH radicals could efficiently activate the CH4molecules.However,the excessive•OH radicals would like to over oxidize the produced oxygenates and lead to a low oxygenates selectivity.In a word,the acquisition and regulation ofin-situgenerated H2O2or•OH radicals are of great importance have been two important challenges in photocatalytic methane conversion.After obtaining sufficient•OH radicals,the amount or concentration ofin-situgenerated H2O2or•OH radicals should be regulated at a proper level.
(4) Advanced characterization technologies are of great significance in understanding activation mechanism of the oxidant as well as CH4.In-situEPR was employed to detect the ROS for insights into the mechanism of O2activation.Operando FTIR and NMR were carried out to investigate the reactive intermediates for the mechanism of CH4activation.Meanwhile,isotope labeling experiments were usually employed to make out the O origination of liquid oxygenates.Specially,to identify the origination of liquid oxygenates products,isotope labeling experiments are necessary in further studies.Therefore,novel characterization technologies should be developed and applied to gain a deeper understanding towards the activation mechanisms of both the selected oxidants and CH4.
(5) An unprecedentedly high CH3OH yield of 12.6 mmol g−1h−1was achieved on Au-Pd/TiO2viaa photothermal synergy.The performance of PPOMO process may be enhanced when coupled with thermocatalysis or electrocatalysis or both.Thus,the synergistic effect should be paid more attention.
In summary,photocatalytic partial oxidation of methane to liquid oxygenates is currently attracting enormous attention.Various research explorations have been expected to soar in the next few years.This review summarized the research progresses in PPOMO from the perspective of the selected oxidants to promote this field step forward and provide new understandings or enlightenments.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
We acknowledge support from the National Key R&D Program of China (No.2021YFA1500800) and National Natural Science Foundation of China (No.22072106).
 Chinese Chemical Letters2023年12期
Chinese Chemical Letters2023年12期
- Chinese Chemical Letters的其它文章
- Spin switching in corrole radical complex
- Benzothiadiazole-based materials for organic solar cells
- Mono-functionalized pillar[n]arenes: Syntheses,host–guest properties and applications✰
- Recent advances in two-step energy transfer light-harvesting systems driven by non-covalent self-assembly✩
- From oxygenated monomers to well-defined low-carbon polymers
- Doping-induced charge transfer in conductive polymers