
保元汤颗粒剂制备工艺和质量控制*
2023-02-14 14:01刘毅刘均正金晨童猛辉陈荷莹郭强黄慧莲
医药导报 2023年2期
刘毅,刘均正,金晨,童猛辉,陈荷莹,郭强,黄慧莲
(1.江西中医药大学现代中药制剂教育部重点实验室,南昌 330004;2.江西和盈药业有限公司,余干 335101)
保元汤收载于2018年国家医药管理局发布的《古代经典名方目录(第一批)》[1],源自《博爱心鉴》[2](明·魏直),由《兰室秘藏》[3](金·李东垣)黄芪汤衍化而来,收录于《简明医彀》[4](明·孙志宏)中,全方由人参、黄芪、甘草、肉桂组成,又加生姜,以水煎服,具有益气、补虚、温阳的功效。现代药理学研究发现保元汤具有抗心力衰竭[5]、抗细胞凋亡[6]、增强免疫能力[7]、保护心肌细胞[8]等作用;临床主要用于治疗冠心病[9],慢性肾炎和肾衰竭[10],再生障碍性贫血[11]和心血管疾病等[12]。传统中药汤剂具有组方灵活、随证加减、起效快、易吸收等特点,但由于缺乏标准的煎煮工艺,如煎煮时间、煎煮温度以及煎液量难以控制,导致汤剂质量不稳定[13]。国家药监局药审中心发布的《按古代经典名方目录管理的中药复方制剂药学研究技术指导原则(试行)》[14]中指出汤剂可制成颗粒剂。相较汤剂,颗粒剂最能维持原方的疗效特点,且具有服用方便、易于携带、能随症加减,故可将汤剂转换为可供快捷、简单、方便使用的剂型[15-16],并通过建立相应质量标准进一步保证其安全性、有效性和可控性。目前文献有关保元汤研究多数处于基础阶段,如古代文献分析[17],化学成分全面定性定量分析[18],标准汤剂和物质基准的指纹图谱及含量测定[19-20],而有关保元汤颗粒剂制备工艺及质量控制研究较少。在质量源于设计(quality by design,QbD)的理念下[21],笔者在本研究采用单因素实验优化辅料及配比,正交设计优化保元汤颗粒剂的制粒工艺;建立物理指纹图谱对制剂的均一性、流动性、堆积性、稳定性进行综合表征;建立高效液相色谱(HPLC)法测定保元汤颗粒剂中指标性成分人参皂苷Rg1、Rb1、毛蕊异黄酮葡萄糖苷和甘草酸铵含量,为制备工艺稳定、质量可控的优质中药颗粒剂奠定基础。
1 仪器与材料
1.1仪器 Sartorius BT25S电子天平(德国赛多利斯公司,感量:0.01 mg);DJTN-2A多功能减压提取浓缩装置(南京至成制药设备有限公司);脉动真空干燥机MZ-0.3m3(常州市震华干燥设备有限公司);LG-20干法制粒机(宜春万申制药机械有限公司);TZ-7型粉体振实密度仪(成都精新粉体测试设备有限公司);D-63150粉末流动性测定仪(德国ERWEKA公司);Agilent 1260高效液相色谱仪[美国Agilent公司,包括四元泵、在线脱气机、自动进样器、DAD检测器和色谱工作站];ACE-C18色谱柱(250 mm×4.6 mm,5 μm,英国ACE公司);ProElut C18-U 500 mg/6 mL 30/pk C18固相萃取小柱(中国迪马科技)。
1.2药材与材料 人参饮片(产地:吉林白山市抚松县,批号:2021090505,人参皂苷Rg1和Re总含量为0.686%,人参皂苷Rb1含量为0.323%);黄芪饮片(产地:甘肃定西市渭源县连峰,批号:LF20171202,黄芪甲苷含量为0.094%,毛蕊异黄酮葡萄糖苷含量为0.064%);甘草饮片(产地:内蒙古阿鲁科尔沁旗,批号:2021051255,甘草苷含量为0.54%,甘草酸含量为3.1%);肉桂饮片(产地:广西防城港市防城区,批号:2021111035,桂皮醛含量为7.4%);生姜药材(产地:江西余干县,批号:20210501,6-姜辣素含量为0.112%,8-姜酚和10-姜方酚总含量为0.041%),以上鉴定均符合《中华人民共和国药典》2020年版1部要求。对照品:人参皂苷Rg1(批号:110703-202034)、人参皂苷Rb1(批号:110704-202129)、毛蕊异黄酮葡萄糖苷(批号:111920-202129)、甘草酸铵(批号:110731-202021),均购自中国食品药品检定研究院。糊精(批号:190808),麦芽糊精(批号:2020030251),二氧化硅(批号:190819),预胶化淀粉(批号:170701),均购自安徽山河药用辅料有限公司。乳糖(荷兰,批号:20141207);微晶纤维素(湖州菱湖新望化学有限公司,批号:20190408);可溶性淀粉(湖州展望药业有限公司,批号:20190630)。
2 方法与结果
2.1保元汤干膏粉的制备 保元汤处方组成为人参3.73 g,黄芪7.46 g,甘草1.86 g,肉桂0.75 g,生姜3 g。称取50倍处方量,置于多功能提取浓缩罐中,以液料比18:1加水,药材浸泡30 min,加热沸腾状态提取60 min;第二次提取液料比13:1,沸腾状态提取60 min。用2层内径0.075 mm(200目)尼龙布滤过,合并两次滤液,70 ℃减压浓缩至清膏(相对密度1.20 g·mL-1),置0.08 kPa,80 ℃的脉动真空干燥箱中干燥,粉碎,过五号筛后备用。
2.2评价指标的测定
2.2.1成型率的测定 按《中华人民共和国药典》2020年版Ⅳ部[22]颗粒剂中粒度项下采用双筛分法操作,以能通过一号筛但不能通过五号筛的颗粒为合格颗粒。
2.2.2吸湿率的测定 称取适量样品于干燥至恒重的称量瓶中,置底部盛有氯化钠过饱和溶液的干燥器中24 h以上,计算颗粒吸湿率[23]。
2.2.3溶化率的测定 按《中华人民共和国药典》2020年版Ⅳ部通则0104颗粒剂项下“溶化性”方法进行测定,计算溶化率。
2.2.4水分的测定 参考《中华人民共和国药典》2020年版Ⅳ部0832水分测定法项的“烘干法” ,计算供试品中含水量(%)。
2.2.5休止角(α)的测定 采用粉末流动性测定仪,选择休止角法,通过漏斗加入保元汤颗粒,记录仪器读数,平行测定3次取均值。
2.2.6相对均齐度指数的测定 将待测颗粒依次通过二、三、四、七、八、九号筛,振荡5 min,分别记录每个筛网截留的颗粒质量。取平均孔径分别为 603,303,188,108,83 μm的筛网截留的颗粒,计算相对均齐度指数。
相对均齐度指数=Fm/[100+(dm-dm-1)Fm-1+(dm-1-dm)Fm+1+(dm-dm-2)Fm-2+(dm+2-dm)Fm+2+…+(dm+n-dm)Fm+n]。
其中,Fm为中间粒径范围粉末的质量百分比,Fm-1为中间粒径范围下一层粉末的质量百分比,Fm+1为中间粒径范围上一层粉末的质量百分比,n为所确定的粒径范围个数,dm为中间粒径范围的粉末平均粒径,dm-1为中间粒径范围下一层的粉末平均粒径,dm+1为中间粒径范围上一层的粉末平均粒径。
2.2.7堆密度(Da)和振实密度(Dc)的测定 按《中华人民共和国药典》2020年版Ⅳ部通则0993堆密度和振实密度测定法测定。
2.2.8豪斯纳比(HR) HR为振实密度与堆密度之比。
2.3辅料的筛选 实验中选择糊精、麦芽糊精、可溶性淀粉等作为辅料,以成型率、振实密度、吸湿率、休止角、吸湿率、溶化率、为评价指标考察辅料种类和用量。结果见表1。
由表1结果可知,药辅质量比2:1和3:2的麦芽糊精成型率较高,但3:2的麦芽糊精溶化过程有少量沉淀,而2:1的麦芽糊精吸湿率、休止角和溶化率均较好,且颜色浅、颗粒均匀度好。综上选择药辅质量比2:1的干膏粉和麦芽糊精进行颗粒剂制备工艺优选。
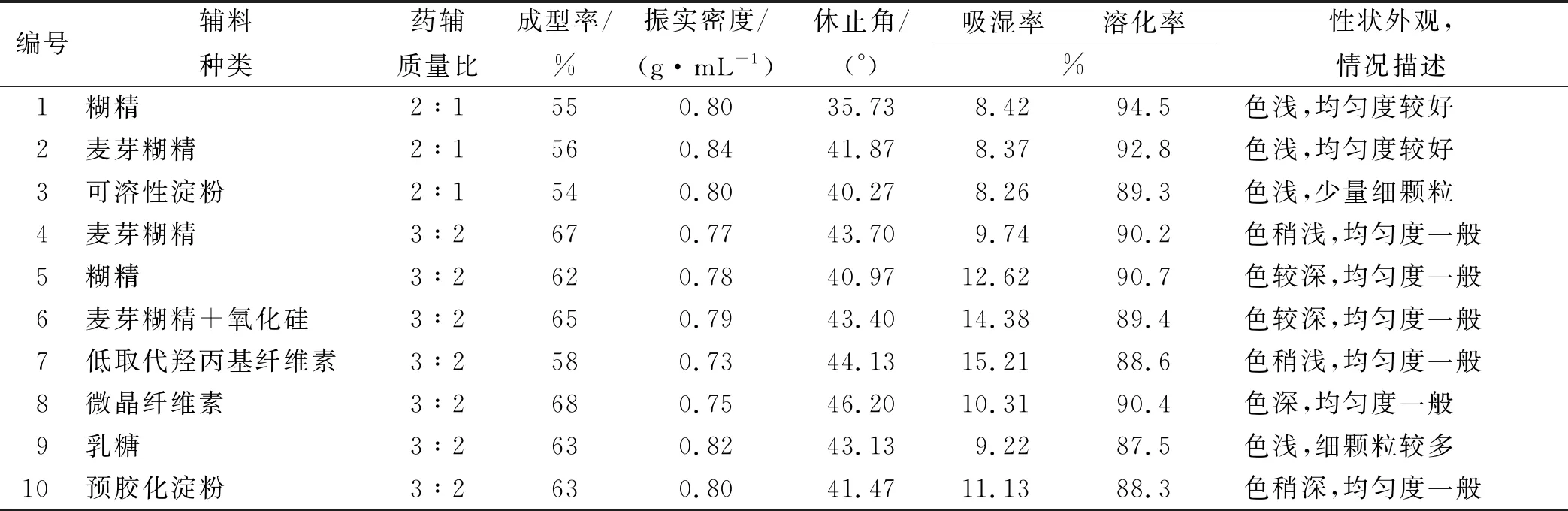
表1 各辅料指标考察结果
2.4正交设计优化颗粒剂制备工艺 干法制粒的主要参数包括辊轮压力,辊轮转速、进料速度;采用正交实验设计助手设计L9(34)正交实验,以成型率为评价指标,考察主要影响因素辊轮压力(因素A)、辊轮转速(因素B)、进料速度(因素C),取药辅比2:1的干膏粉和麦芽糊精进行实验。正交实验因素水平表见表2。正交实验结果见表3,方差分析结果见表4。

表2 正交实验因素水平表
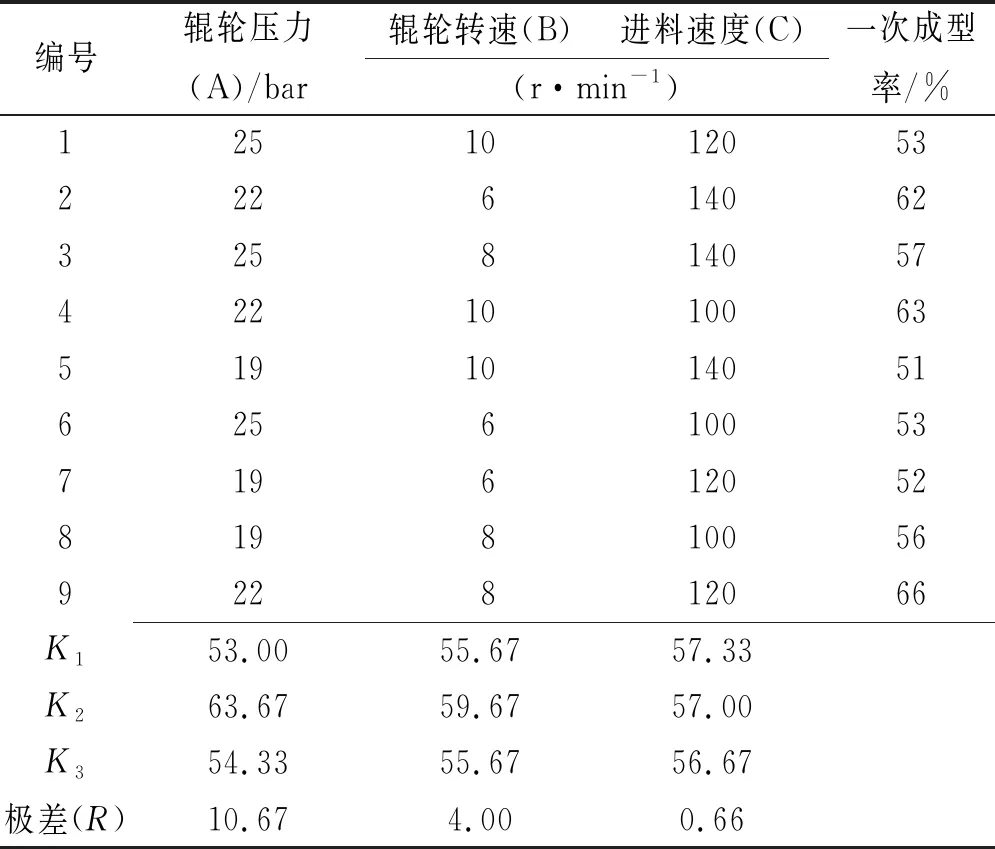
表3 L9(34)正交实验结果
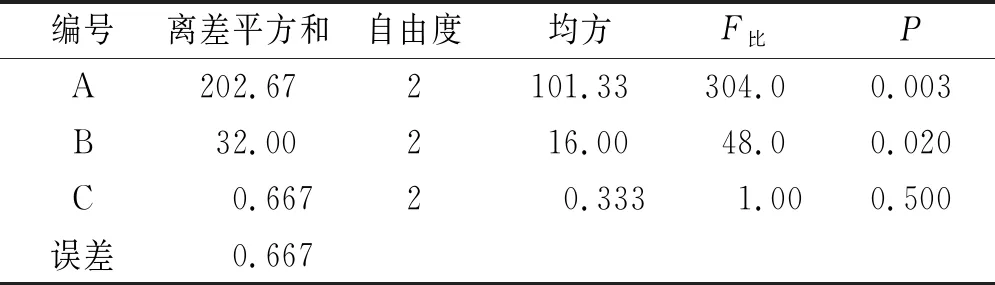
表4 方差分析结果
方差分析可知辊轮压力对干法制粒有极显著影响(P<0.01),辊轮转速具有显著差异(P<0.05),进料速度(P>0.05)对颗粒剂制备无明显影响。表明辊轮压力对颗粒剂制备的影响最大,其次是辊轮转速,进料速度的影响较小。综上可知最佳工艺参数为辊轮压力22 bar,辊轮转速8 r·min-1,进料速度100 r·min-1。
2.5验证性实验 按照以上筛选的处方配比和优化的最佳工艺进行五批验证性实验的考察,以验证本工艺的稳定性。结果显示,五批验证实验的成型率、溶化率和色泽外观等结果相接近,说明该法制备工艺稳定可行,结果见表5。
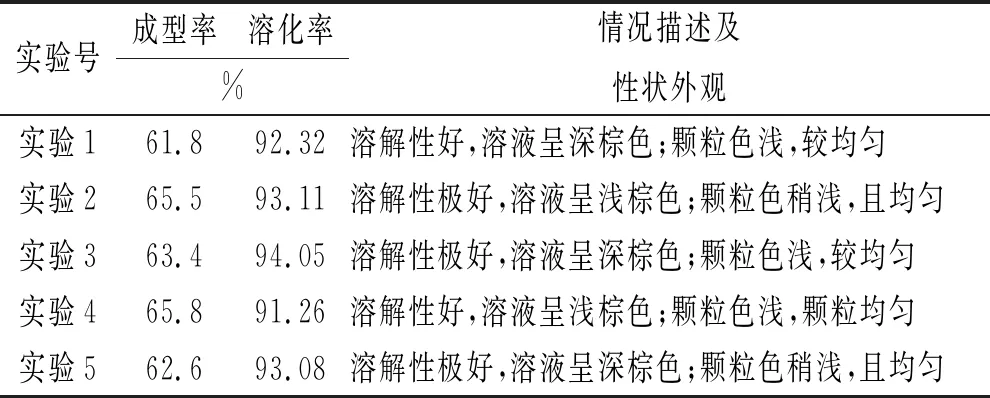
表5 5批验证实验结果
2.6物理指纹图谱的建立 物理指纹图谱可从物理性质层面评价颗粒剂质量,参考文献[24-26],设计4个一级指标和7个二级指标。一级指标可表征颗粒剂的性能或功能,二级指标是一级指标的具体参数,可表征均一性、堆积性、稳定性和流动性[27],依据此建立物理指纹图谱,评价不同批次颗粒剂质量的一致性。
2.6.1物理指标的确定及标准化转换 由于二级物理质量指标的量纲不同,直接分析会影响评价结果的准确性,为方便图谱展示,可对各二级指标进行标准化或归一化处理,以便在同一尺度上对不同指标进行展示。指标转换方法参考药用辅料手册和《欧洲药典》标准[28],先查找每个质量指标上下限数值范围,将其数值标准化至同一尺度0~10,公式见表6,结果见表7。
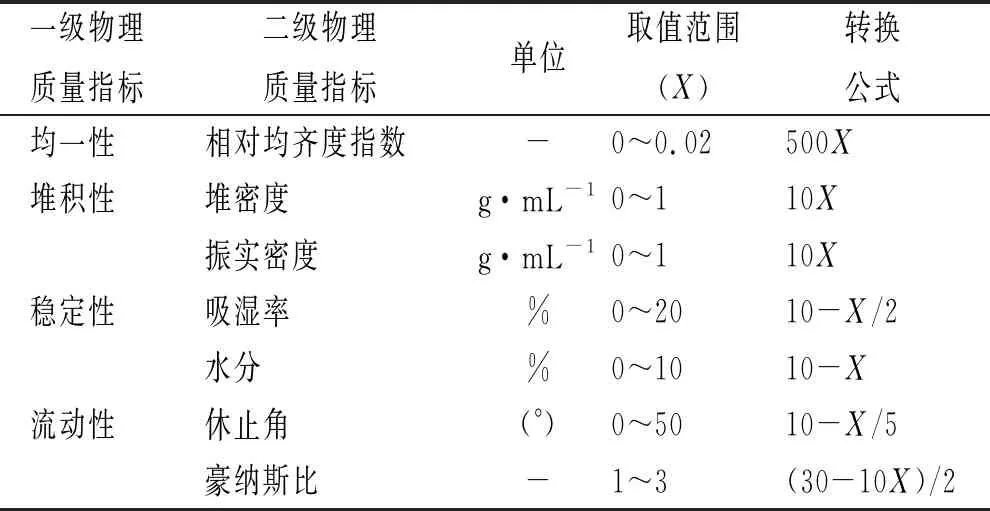
表6 各指标的标准转换公式
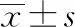
表7 二级物理指标测定结果
2.6.2物理指纹图谱的构建及相似度分析 雷达图可以定量直观的展示出各指标之间关系,即可视化的颗粒物理指纹图谱。将多批次样本测定的各指标数据综合处理后,分别以二级指标绘制雷达图作为颗粒剂物理指纹图谱,以12点钟方向为起始点按顺时针方式依次排列,有色多边形区域表示保元汤颗粒的物理指纹图谱。结果表明验证1~5与对照指纹图谱比较相似度均>0.99,5批验证颗粒剂的物理质量与对照颗粒剂一致。物理指纹图谱见图1。
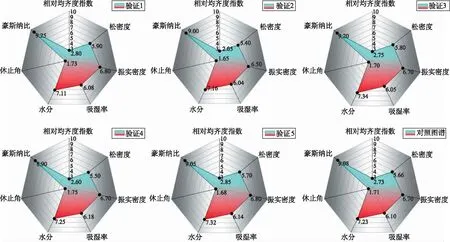
图1 颗粒剂物理指纹图谱
2.7保元汤颗粒剂的含量测定
2.7.1色谱条件 采用ACE-C18柱(250 mm×4.6 mm,5 μm)色谱柱;柱温:25 ℃;流速:1.0 mL· min-1;检测波长:254 nm(毛蕊异黄酮葡萄糖苷、甘草酸铵),203 nm(人参皂苷Rg1、人参皂苷Rb1);流动相:乙腈和0.1%磷酸水溶液,按表8梯度洗脱。

表8 梯度洗脱程序表
2.7.2混合对照品溶液的制备 精密称定毛蕊异黄酮葡萄糖苷2.99 mg,加甲醇定容至10 mL量瓶,超声混匀备用;精密称定人参皂苷Rg16.09 mg、人参皂苷Rb15.38 mg、甘草酸铵5.41 mg,精密吸取以上毛蕊异黄酮葡萄糖苷2.5 mL置于同一5 mL量瓶中,加甲醇定容,制成混合对照品溶液,浓度分别为:毛蕊异黄酮葡萄糖苷149.5 μg·mL-1、人参皂苷Rg11218 μg·mL-1、人参皂苷Rb11076 μg·mL-1、甘草酸铵1082 μg·mL-1。
2.7.3供试品溶液的制备 精密称取颗粒剂研细粉末0.5 g,加20%甲醇10 mL溶解,超声30 min,4 000 r·min-1离心10 min,吸取全部上清液,加在C18固相萃取小柱上,吸附后,用双蒸水和15%甲醇各10 mL洗脱,弃去;再加纯甲醇洗脱并收集,洗脱液定容至5 mL容量瓶中,摇匀,滤过,即得。
2.7.4阴性供试品溶液的制备 按照优化后的最佳工艺制备分别缺少人参、黄芪、甘草的阴性颗粒剂,再按照“2.7.3供试品溶液的制备”方法制备阴性供试品溶液。
2.7.5专属性研究 取混合对照品、供试品和阴性供试品溶液,按照“2.7.1”项色谱条件分别进样,选择203和254 nm双波长记录色谱图,见图2(254 nm)和图3(203 nm)。结果显示,所有缺单味药的阴性供试品溶液在测定成分出峰时间处均无干扰,各成分之间的分离度良好,分析方法可行性强,表明方法专属性良好。

A.混合对照品;B.供试品;C.黄芪阴性供试品;D.甘草阴性供试品.1.毛蕊异黄酮葡萄糖苷;2.甘草酸铵。
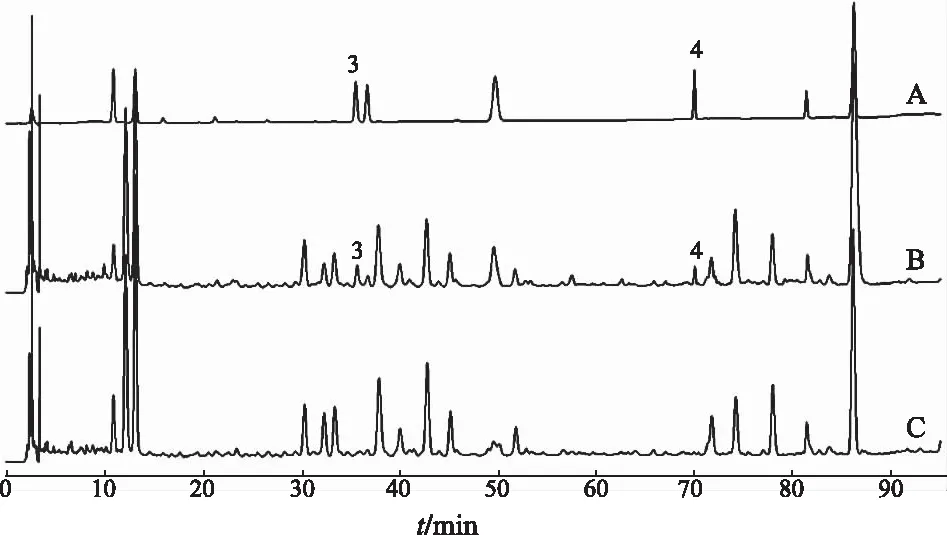
A.混合对照品;B.供试品;C.人参阴性供试品.3.人参皂苷Rg1;4.人参皂苷Rb1。
2.7.6线性关系考察 精密吸取 “2.7.2”项下制备的混合对照品溶液1 mL,以甲醇逐级稀释至2 mL量瓶中,配制成不同浓度的对照品溶液,分别吸取10 μL注入高效液相色谱仪进行测定,每个样品连续进样2针,在254和203 nm波长下记录峰面积,分别以对照品浓度为横坐标,对应峰面积为纵坐标,绘制标准曲线,得到各成分线性回归方程和线性范围,结果见表9。

表9 保元汤中4种化学成分的线性关系
2.7.7精密度考察 取“2.7.2”项下混合对照品溶液稀释3倍后的溶液,按照“2.7.1”项下色谱条件连续进样6次。结果表明:毛蕊异黄酮葡萄糖苷、人参皂苷Rg1、人参皂苷Rb1、甘草酸铵峰面积的RSD值分别为0.25%、0.37%、0.70%、0.77%,均<1.0%,说明该方法的精密度良好。
2.7.8稳定性考察 精密称定颗粒剂粉末约0.5 g,按照“2.7.3”项下方法制备供试品溶液,按照“2.7.1”项下色谱条件进样,分别于0,6,12,18,24,30,36 h进样7次。结果表明:毛蕊异黄酮葡萄糖苷、人参皂苷Rg1、人参皂苷Rb1、甘草酸铵峰面积的RSD值分别为0.74%、2.22%、1.29%、0.36%,均<3.0%,说明供试品溶液在36 h以内稳定。
2.7.8重复性考察 精密称定来源于同一批次颗粒剂粉末的供试品6份,每份约0.5 g ,按照“2.7.3”项下方法制备6份供试品溶液,按照“2.7.1”项下色谱条件进样,记录色谱图,分别计算供试品中各成分的含量和RSD值,考察结果表明:毛蕊异黄酮葡萄糖苷、人参皂苷Rg1、人参皂苷Rb1、甘草酸铵含量的RSD值分别为2.52%、2.52%、0.95%、3.00%,均<3.0%,说明供试品溶液的制备方法稳定,重复性好。
2.7.9加样回收率考察 精密称定来源于同一批次颗粒剂粉末的供试品6份,每份约0.25 g,置于具塞锥形瓶中,分别精密加入对照品适量,按照“2.7.3”项下方法制备6份供试品溶液,按照“2.7.1”项下色谱条件进样,记录色谱图,计算供各成分的加样回收率和RSD值,结果显示供试品中各指标性成分的平均回收率介于96.35%~105.83%,均符合要求,见表10。
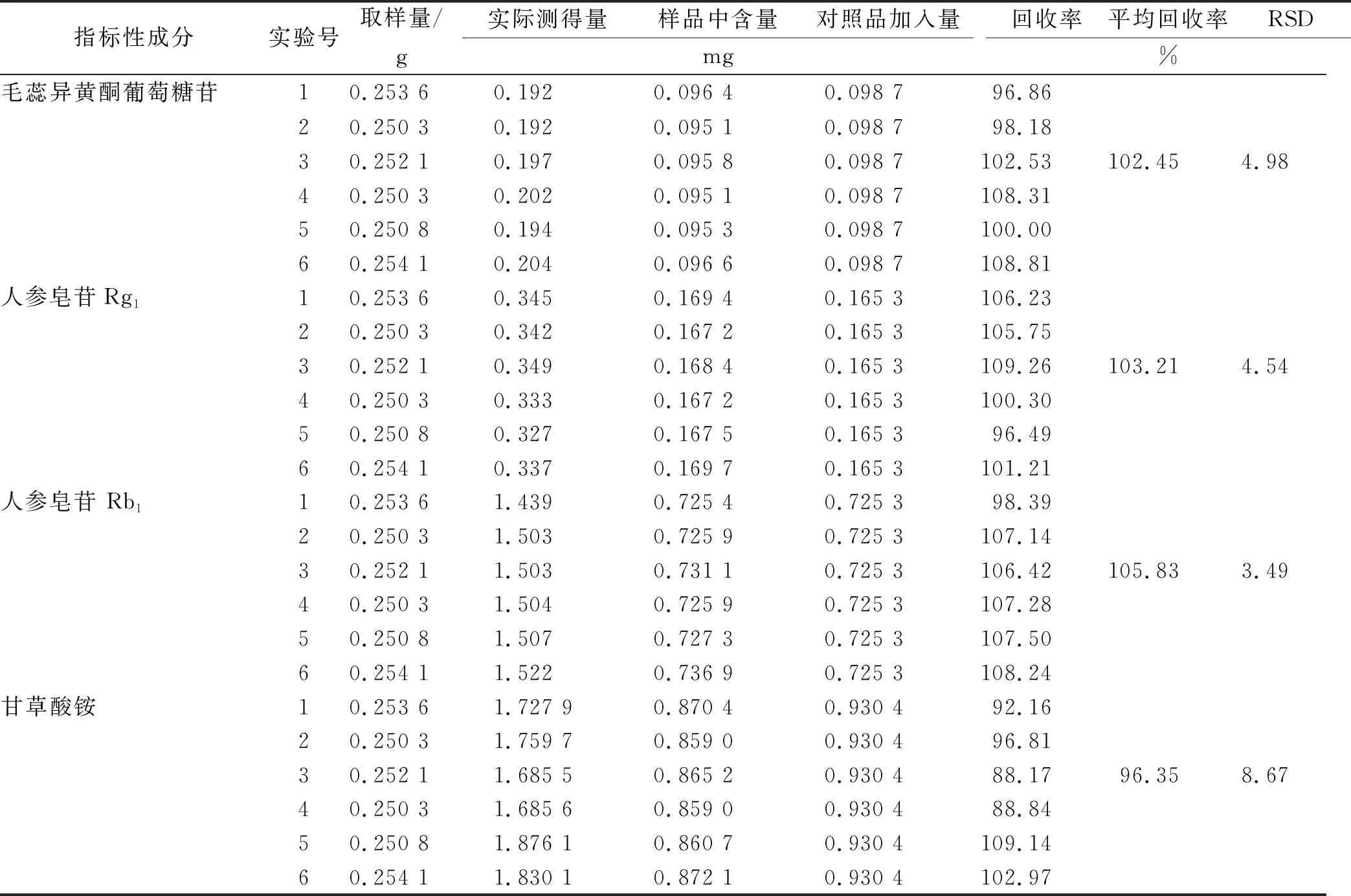
表10 供试品中各成分加样回收率测定结果
2.7.10样品的含量测定 按“2.7.3”项方法制备5批次保元汤颗粒剂供试品,按照“2.7.1”项色谱条件进样,计算单个处方中各指标性成分的含量,结果见表11。
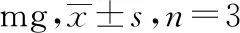
表11 保元汤颗粒剂含量测定结果
3 讨论
中药复方具有成分多样且复杂、中间体易吸潮、黏性大等特点,制粒过程应当充分考虑颗粒的流动性、飞散性、黏附性,还应保证成型颗粒的形状大小均匀、外形美观便于准确计量,因此应当根据不同处方中的成分差异,选择不同的制粒方法以及不同设备。翟红伟等[29]采用湿法制粒制备保元颗粒,将浸膏进行喷雾干燥后,选择麦芽糊精为辅料,麦芽糊精与干膏粉用量比例为1:1,以90 %乙醇为湿润剂,进行湿法制粒后,再干燥;可见湿法制粒操作过程步骤较多,生产过程中浸膏和颗粒均需要干燥,多次干燥且时间较长可能会破坏或污染有效成分。而干法制粒对有效成分影响小,操作简单,更适宜对湿、热敏感性高的药物颗粒制备,还能省去制软材和干燥过程,具有较大优势,可减少辅料使用量从而提高载药量[30]。
本研究首次对保元汤汤剂采用干法制粒,直接将干膏粉与适宜辅料混合后制粒,无需干燥,操作简单方便,适用于现代大生产。该处方中人参与黄芪占比较大,苷类成分较多致使干膏粉吸湿性较强,为减小其吸湿性,增加颗粒剂顺应性,需要加入适量辅料,辅料种类和用量直接影响颗粒成型的结果,还影响患者服用剂量的多少。本研究先对多种辅料的筛选和药辅质量比的考察,综合得出药辅质量比2:1的麦芽糊精各评价指标较好。正交实验确定保元汤干法制粒工艺为辊轮压力22 bar,辊轮转速8 r·min-1,进料速度120 r·min-1。采用雷达图构建颗粒的物理指纹图谱,可直观快速判断颗粒剂成型后物理性质的可能缺陷,并通过在制剂处方中增加相应辅料予以改良。通过SPSS17.0版软件的夹角余弦法比较不同批次间样品物理指纹图谱与对照指纹图谱的相似度,相似度越大,越接近1,表明颗粒剂的物理性质越相似,颗粒剂质量越稳定。
制粒过程发现,物料性质是影响干法制粒的关键因素,一方面,物料若是含糖类成分,易受环境的影响,在空气中易吸潮,致使粘度增大,增加了制粒难度;另一方面,制粒机连续工作时,滚轮间温度升高,粉末可溶性增加,致使滚轮与粉末之间黏附力增加,导致颗粒成型率降低。因此,制粒仪器的性能、环境温湿度和辅料种类及用量的保障是干法制粒的关键条件;笔者建议首先应对干膏粉物理性质综合考察,依据此选择适宜辅料及药辅比例,最后确定适合的制粒方法和仪器设备;制粒过程中需要选择合适的环境以减少温湿度的干扰。
猜你喜欢
现代农药(2022年4期)2022-11-19
安徽农业大学学报(2022年2期)2022-11-09
世界科学技术-中医药现代化(2021年5期)2021-11-05
纺织服装流行趋势展望(2020年1期)2020-02-01
科学之谜(2018年3期)2018-04-09
新型建筑材料(2018年2期)2018-03-09
纺织服装流行趋势展望(2016年6期)2016-05-04
纺织服装流行趋势展望(2016年4期)2016-05-04
纺织服装流行趋势展望(2016年1期)2016-05-04
家庭科学·新健康(2015年6期)2015-06-04
