
中兽药口服液中非法添加化学药物高分辨质谱筛查
2023-01-24 09:50霍宁宁李研东艾连峰王振来
分析测试学报 2023年1期
霍宁宁,李研东,韩 雪,艾连峰,王振来
(1.河北科技大学 食品与生物学院,河北 石家庄 050018;2.河北省兽药饲料工作总站,河北 石家庄 050035;3.石家庄海关技术中心,河北 石家庄 050011)
农业农村部2019年7月发布的194号公告[1]中规定,除中药、驱虫药等部分药物外,养殖环节禁止使用所有促生长和治疗预防性药物。自此药物饲料添加剂退出历史舞台。而中兽药因其天然、毒副作用小、价格低廉、符合法规要求等优势[2],在兽药市场的激烈竞争中脱颖而出。中兽药产品的快速崛起,在带来利润的同时也给不法分子带来了可乘之机,有些企业利用中兽药检测技术滞后单一、标准匮乏的现状,通过添加化学药物的手段达到非法获利的目的[3]。中药植物源性成分来源多样,有效成分复杂,如生物碱类、黄酮类、皂苷类、色素、糖类、挥发油等物质均可对常规仪器检测产生影响[4-5],因而快速筛查其中的非法添加物十分困难,且非法添加药物的广泛性和隐蔽性也对检测仪器的选择性、分辨率和准确性提出更高要求,传统兽药检测方法和仪器显然无法满足高通量和快速的检测要求。目前报道的关于兽药中违禁药物添加方面的仪器检测技术主要有分光光度法[6]、气相色谱法[7]、液相色谱法[8-9]、质谱法[2,10-11]等。其中《中华人民共和国兽药典》[12]和农业农村部公告[13]等规定的兽药检验方法多以液相色谱法为主,其前处理过程往往依据现有药典和标准规范,方法适用性较差,抗干扰能力不足,检测的目标物有限,对于兽药中违禁药物的检测局限性较大。辜慧等[3]建立了UPLCMS∕MS法同时检测减肥类、壮阳类、降糖类、解热镇痛类中药制剂中易被添加的27种化药成分,但检测种类少,且此种类的中药制剂成分较为简单;曹秀等[10]建立了26种兽药制剂中喹诺酮类、磺胺类、孔雀石绿及金刚烷胺的检测方法,检出限低,但检测药物种类少,在筛查中可能会忽略其他非法添加药物;林仙军等[14]采用UPLC-Q-TOF MS法建立了快速筛查兽药中72种非法添加化学药物的方法,但药物的添加浓度较高。目前关于兽药中非法添加化合物的分析标准只有四川省的DB 51∕T 1855-2014[15]和山东省的DB 37∕T 2817-2016[16],而相关的国家和行业标准均未见报道。
随着高分辨质谱的应用日益广泛,其高选择性、高灵敏度、高分辨率,以及对未知化合物的高通量筛查优势愈加凸显。结合高速发展的样品前处理技术和复杂样品基质成分的多组分分析技术被更多的研究者采用[14,17-24],本文采用0.5%甲酸乙腈-水对样品进行提取,将提取液经Oasis PRiME HLB固相萃取小柱净化后,超高效液相色谱-四极杆静电场轨道阱质谱进行检测分析,建立了小分子筛查数据库,结合Trace Finder筛查软件分析,初步确立了适用于中兽药口服液中167种非法添加药物的高通量快速筛查方法。该方法可进行上百种非法添加药物的筛查检测,适用于复杂中药基质的高选择性、高通量的筛查,一定程度上扩充了药物筛查种类和数量,适用于具备仪器条件的政府监管部门和第三方检测机构使用,也可为相关企业在原材料质量风险控制、成品监督等方面提供借鉴。
1 实验部分
1.1 试剂与仪器
167种兽药标准品(First Standard,阿尔塔公司);乙腈(色谱纯,默克公司);屈臣氏蒸馏水;甲酸(88%,赛默飞世尔科技公司)。
UltiMate 3000-Q-Exactive Focus超高效液相色谱-四极杆静电场轨道阱高分辨质谱,Accucore RP-MS色谱柱(Thermo公司);Oasis PRiME HLB(6 mL,200 mg)固相萃取柱、HLB(3 mL,60 mg)固相萃取柱、MCX(6 mL,150 mg)固相萃取柱(Waters公司);Captiva EMR-Lipid(3 mL,300 mg)固相萃取柱(Agilent公司)。
1.2 标准溶液配制
167种质量浓度为100 µg∕mL的标准品混标溶液,配制成10 µg∕mL的混标标准储备液;准确量取167种混标储备液(10 µg∕mL),用甲醇稀释成1 µg∕mL的混合标准工作液待用。
1.3 仪器条件
液相色谱条件:色谱柱:Accucore RP-MS柱(100 mm × 2.1 mm,粒径2.6 µm);流动相A:0.05%甲酸水;流动相B:0.05%甲酸乙腈;梯度洗脱程序:0 ~ 2.5 min,98% ~ 95% A;2.5 ~ 5.8 min,95% ~90% A;5.8 ~ 6.5 min,90% ~ 85% A;6.5 ~ 7.5 min,85% ~ 80% A;7.5 ~ 9.5 min,80% ~ 70% A;9.5 ~10.5 min,70% ~ 60% A;10.5 ~ 12 min,60% ~ 55% A;12 ~ 14 min,55% ~ 45% A;14 ~ 18 min,45% ~5% A;18 ~ 21 min,5% A;21 ~ 22.5 min,5% ~ 98% A;22.5 ~ 25 min,98% A;流速:0.3 mL∕min;进样量:5 µL;柱温:30 ℃。
质谱条件:采用电喷雾离子源(ESI源),正负离子检测模式(ESI+、ESI-)同时扫描,扫描方式为一级全扫描,数据依赖型二级质谱扫描(Full MS∕dd-MS2),源区主要参数设置:鞘气流速:9 arb,喷雾电压:3 kV,毛细管(离子传输管)温度:320 ℃,S-lens电压:55 kV,辅助气加热温度:30 ℃。质谱一级扫描模式为Full MS,扫描范围(m∕z)为100 ~ 900,一级分辨率为70 000,C-Trap的离子数目为1 × 106,离子最大注入时间为100 ms;二级扫描模式为dd-MS2模式,二级分辨率为35 000,顶点激发时间为3 ~ 6 s,设置阶梯碰撞能分别为20、35、50 kV,动态排除时间为8 s。采集数据前使用正负离子校正液进行质量轴校正。质谱采集数据通过Xcalibur质谱工作台提取化合物碎片离子信息,通过Trace Finder筛查软件建立筛查数据库。
1.4 样品前处理
为构建适用于绝大部分样品的前处理方法,实验选择样品基质成分复杂,且不容易去除基质影响的清解合剂做为代表性样品。
提取:称取中兽药口服液样品0.25 g(精确至0.01 g),置于50 mL离心管中,准确加入20 mL 0.5%甲酸乙腈水(乙腈∶水 = 9∶1)溶液,涡旋混匀10 min,超声提取20 min,12 000 r∕min 离心10 min;取上清液至25 mL容量瓶中定容,待净化。
净化:取上清液5 mL,过Oasis PRiME HLB固相萃取小柱,将流出液在40 ℃下氮吹浓缩至0.1 mL左右,用0.2%甲酸水定容至1 mL,涡旋混匀做为原液。取原液0.2 mL定容到1 mL,涡旋混匀后做上机液,上机液过0.2 µm微孔滤膜后,供质谱分析。
1.5 筛查结果判定
参考《兽药残留检测中质谱方法确证指南》[25]和欧盟2002∕657∕EC[26]准则,应用高分辨质谱对目标化合物进行分析时,若有1个母离子和1个子离子或2个子离子匹配则可确证分析的样品含有该化合物。对于疑似阳性样品,进一步在dd-MS2模式下针对疑似含有的化合物进行二级特征碎片离子扫描,当有2个或以上丰度较高的碎片离子与标准谱图中相应的碎片离子精确质量偏差小于5 ppm且碎片离子的相对丰度比在最大允许偏差范围内时,确证含有该化合物。
对供试品溶液进行Full MS∕dd-MS2全扫描,根据建立的标准筛查数据库中m∕z的精确分子质量、保留时间和至少有一个碎片离子对目标化合物进行筛查。当未知物出峰时间偏差在 ± 0.5 min内,母离子响应值在105及以上、精确质量数偏差在5 ppm范围内,子离子响应值在104及以上、精确质量数偏差在5 ppm范围内,同位素丰度比范围在70%范围内,则检测结果为疑似阳性样品,否则判定为阴性样品。
2 结果与讨论
2.1 筛查数据库的建立
根据167种非法添加化学药物的标准品信息,通过仪器方法分析,自建了167种化合物的基本信息及色谱-质谱信息数据库(见表1)。

表1 167种化合物的基本信息及色谱-质谱信息Table 1 Basic information and chromatography-mass spectrometry information of 167 compounds

(续表1)
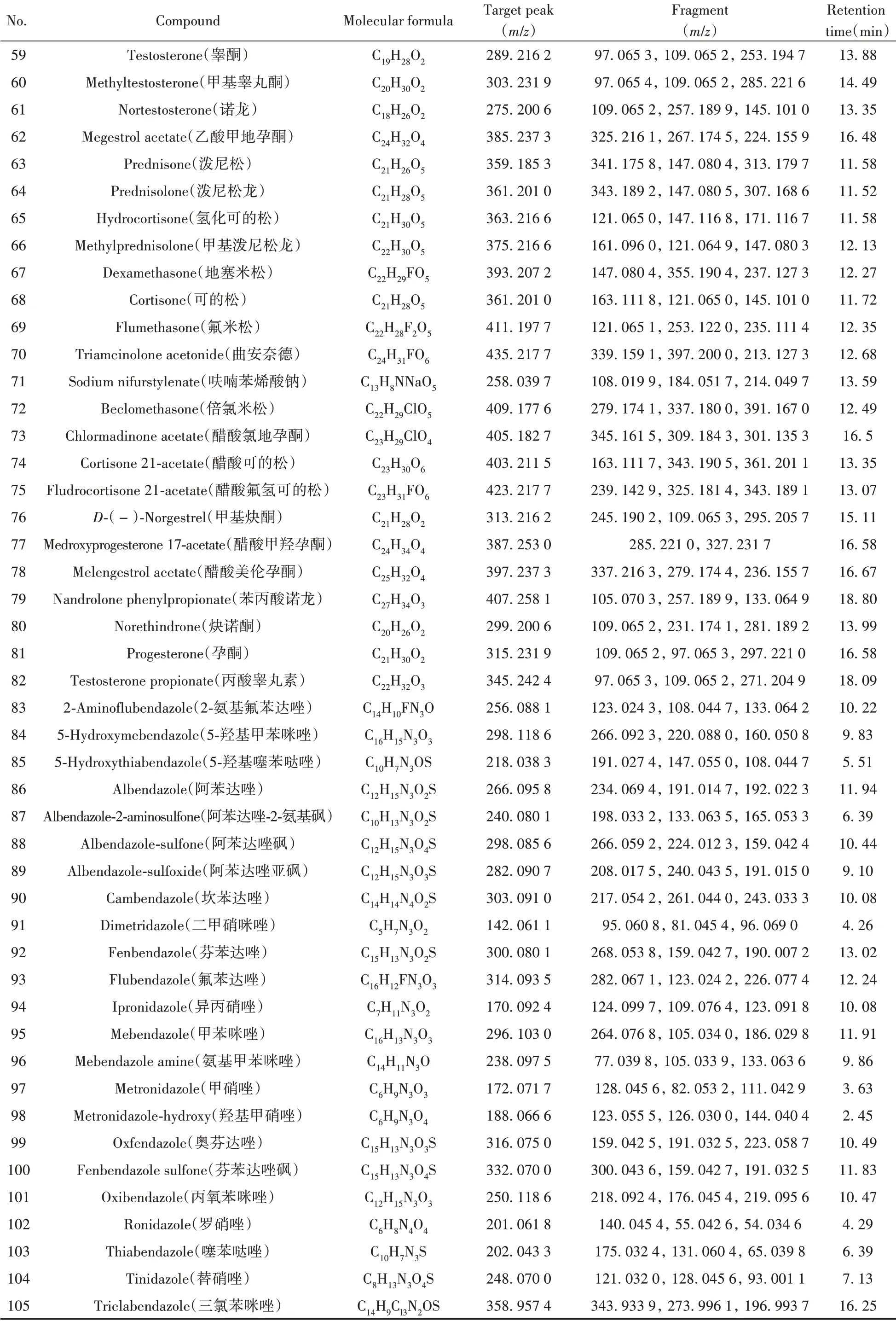
(续表1)
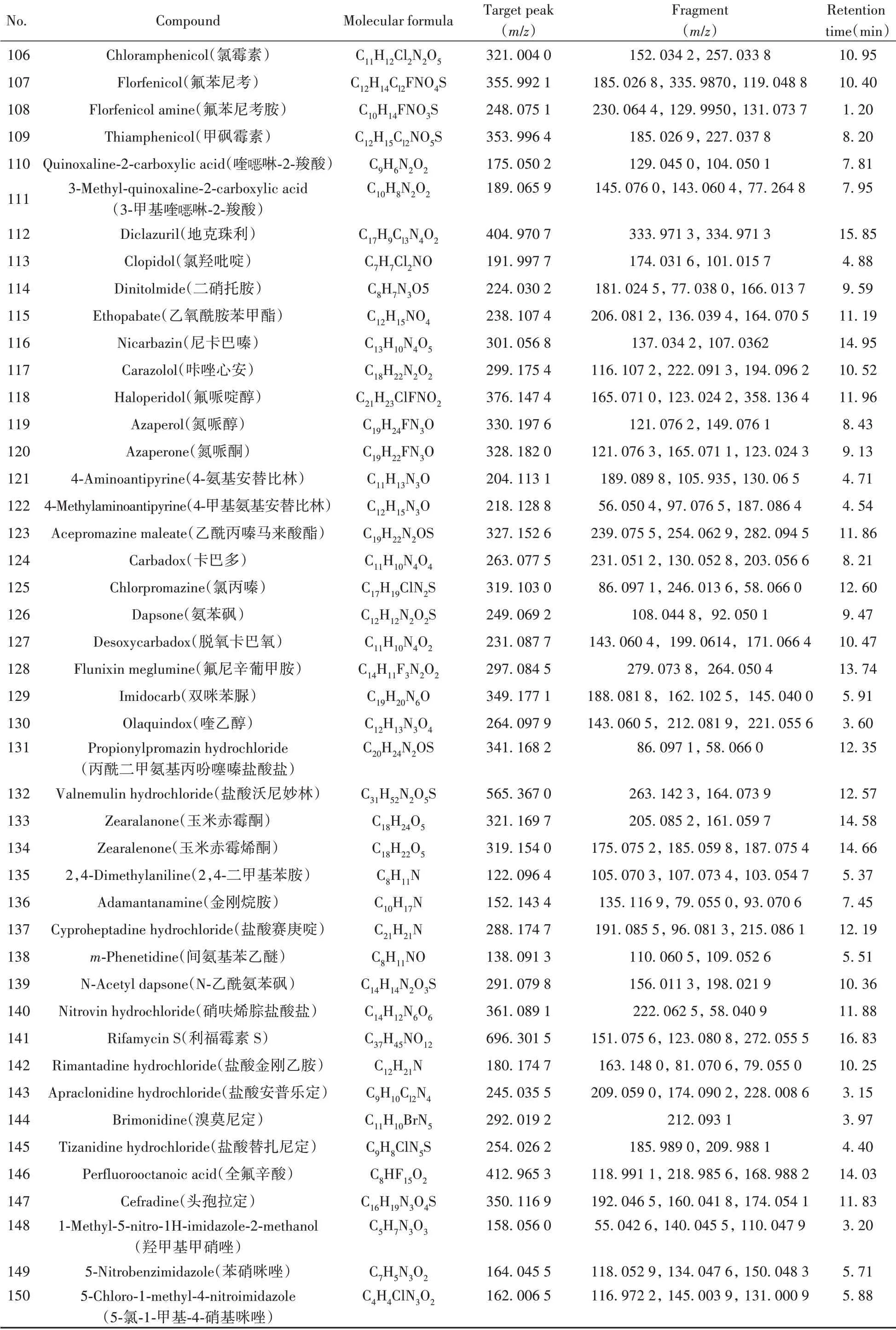
(续表1)
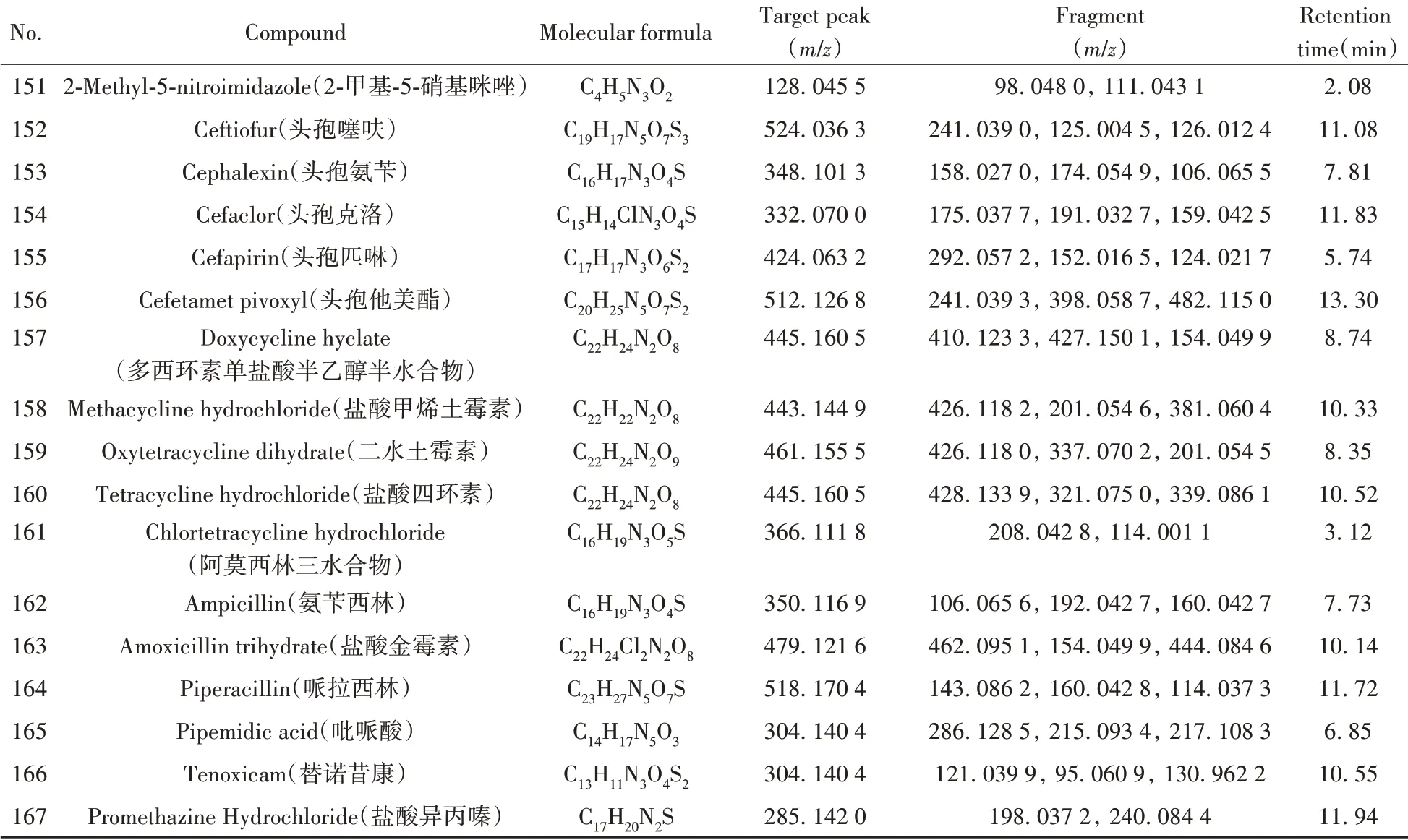
(续表1)
2.2 色谱条件的优化
考察了多个因素对色谱分离、灵敏度及离子化效率的影响,包括流动相及流动相梯度选择。由于本文所考察的化合物性质差异较大,含有8对同分异构体,多西环素单盐酸半乙醇半水合物与盐酸四环素,泼尼松龙与可的松,磺胺间二甲氧嘧啶与磺胺邻二甲氧嘧啶,盐酸莱克多巴胺与盐酸苯氧丙酚胺,磺胺氯哒嗪与磺胺氯吡嗪,磺胺对甲氧嘧啶、磺胺甲氧哒嗪与磺胺间甲氧嘧啶,磺胺二甲基嘧啶与磺胺二甲异嘧啶,磺胺恶唑与磺胺异恶唑,本实验首先对流动相的梯度条件、流动相的成分进行考察,以实现目标化合物的最佳分离以及各化合物的良好峰形和最佳响应。考察了乙腈-0.05%甲酸水、0.05%甲酸乙腈-0.05%甲酸水、甲醇-水和乙腈-水分别作为流动相时,不同梯度下目标物在Accucore RP-MS(100 mm × 2.1 mm,2.6 µm)色谱柱上的分离效果。结果显示,在甲醇-水或乙腈-水中,同分异构体均未能完全分离,与甲醇-水相比,乙腈-水中均加入0.05%甲酸时,增加了化合物的电离效果,大部分待测物的相应灵敏度提高,峰形得到改善;理论上,加入0.1%甲酸时化合物的电离效果会更加明显,但对于负离子模式的化合物而言,会抑制负离子模式的化合物电离。考虑到目标物在正负离子模式所需的最适溶液pH和当前的灵敏度和峰形均已满足要求,且上述8对同分异构体均已实现完全分离(见图1),因此实验最终选择0.05%甲酸乙腈-0.05%甲酸水作为最佳流动相,并按“1.3”梯度洗脱条件进行分离。

图1 8对同分异构体的分离色谱图Fig.1 Separation chromatograms of 8 pairs of isomers
2.3 质谱条件的优化
对扫描模式、碰撞能、采集点数和动态排除时间进行优化。在选择扫描采集模式中,根据167种化合物的化学性质,全氟辛酸、氯霉素类和地克珠利以[M - H]-的响应值最高,其他化合物以[M + H]+的响应值最高。由于仪器可提供正、负模式同时扫描,本实验选择正、负模式同时扫描对167种化合物进行采集。通常进行二级质谱数据库的检索时,低碰撞能量的质谱图有利于判断主要碎片,高碰撞能量的质谱图可得到更多质谱碎片细节,提高化合物的定性准确性,由于实验涉及的化学物质种类繁多,本文设置了较宽范围的分步碰撞能,以获得最适合的碰撞能击打碎片。在质谱扫描采集点数优化过程中,质谱采集点数以12 ~ 15个为最优,且Chorm peak Width和Smoothing中的Points参数值变大可改善质谱正负模式同时扫描中采集点数不够的问题,因此实验将Chorm peak Width参数由15改为30,将Smoothing 中的Points数值从3增至7,以增加采集点数,此时色谱峰形逐渐平滑且趋于正态分布而非平头峰趋势(如图2)。为避免重复采集响应强度占优势的某一质荷比的母离子的二级质谱,尽可能获得不同质核比的母离子的二级谱图,本实验将动态排除时间缩短(由15 s改为8 s),以增加获得不同质荷比母离子的二级质谱信息的机率,避免出现漏采集情况。

图2 谱图采集点数的比较Fig.2 Comparison of the number of spectral acquisition points
2.4 提取溶剂的选择
由于本实验涉及化合物的极性差异较大,且兽药基质多为水溶性杂质。在参考文献的基础上,考察了甲醇、乙腈、0.1%乙酸乙腈、1%乙酸乙腈、0.5%甲酸乙腈、2%甲酸乙腈、0.5%甲酸乙腈-水(9∶1)作为提取液的提取效果。结果显示(见图3),在提取液中加入适当酸可提高化合物的筛查检出率,且甲酸的效果优于乙酸,但过量的酸度会抑制硝基咪唑类、头孢类、内酰胺类物质的提取效果。就提取液的颜色而言,0.5%甲酸乙腈提取的样品溶液颜色较浅,表明色素含量少。在实验过程中,因为口服液本身含有水分,考虑到待测化合物为水溶性化合物,故在提取液中加入少量水可提高筛查检出率,满足实验要求。本实验最终选择0.5%甲酸乙腈-水(9∶1)为提取液。

图3 不同提取液对添加药物的筛查结果Fig.3 Screening results of different extract soltions for drug addition
2.5 净化条件的优化
为有效去除酮、醛、酚等干扰物质的影响,增加对靶标物质定性和定量检测的准确性,本研究参考畜产品的前处理过程,考察了4种不同品牌和填料的固相萃取小柱(PRiME HLB固相萃取柱、HLB固相萃取柱、EMR固相萃取柱、MCX固相萃取柱)的分离、净化和富集效果。结果显示,PRiME HLB和EMR固相萃取柱无需活化、平衡等步骤,可将提取液直接过柱达到净化目的,但后者需采用比例为4∶1的提取液-水上样;而HLB和MCX固相萃取柱需经过一系列活化上样,过程较繁杂。此外,PRiME HLB固相萃取柱对绝大多数化合物的保留性强,洗脱液颜色相较提取液明显变浅,原因可能为部分色素、生物碱、皂苷等保留在净化柱上。进一步考察了上样体积的影响,结果显示,若上样体积过低,则待测物可能因洗脱体积小而保留在净化柱上;加大上样体积,则杂质目标物同时洗脱出固相萃取柱,导致检出效果变差。最终选择5 mL提取液过PRiME HLB固相萃取柱净化,以获得最好的实验效果。
2.6 样品分析
考察显示,167种药物的标准溶液在上机质量浓度为20 ng∕mL时,绝大部分药物均检出,头孢类和内酰胺类药物在50 ng∕mL时检出。因基质影响,不同药物配方非法添加药物的检出率略有差异,配方成分复杂的口服液(如:清解合剂、清瘟解毒口服液等)前处理后,药物检出浓度高于简单配方的中药口服液。如四逆汤样品以50 ng∕mL上机时绝大部分药物均可检出,头孢他美酯、头孢匹啉、氨苄西林和哌拉西林等不能检出的头孢类药物和内酰胺类药物,可在100 ng∕mL上机浓度时检出;选择含石膏、金银花、玄参、黄芩、生地黄、连翘、栀子和龙胆等12种中药基质成分的清解合剂空白样品为基质,非法添加药物在50 ng∕mL水平下(头孢类和内酰胺类药物为100 ~ 200 ng∕mL)可定性检出,其中药物上机质量浓度为100 ng∕mL时,检出率为95.81%,药物上机质量浓度为200 ng∕mL时,药物检出率可达100%。依据违法者在中兽药中主观添加非法药物发挥抗病或治疗效果的目的,本文的检测浓度已满足实际检测的需要。
3 结 论
本文基于酸性有机溶剂振荡超声提取的方式,对不同中兽药口服液中的167种非法添加化学药物进行快速测定;采用自建的筛查数据库比对样品数据结果,检出率可满足分析测定要求;使用UltiMate 3000-Q-Exactive Focus进行分析,精确测得目标物母离子及子离子的分子量、保留时间,降低了测定结果的不准确性。该方法采用Trace Finder筛查软件建立了167种非法添加化学药物的高分辨质谱数据库,可有效实现不同中兽药基质中非法添加化学药物的快速检测,满足对中兽药口服液中非法添加物定性筛查的要求,为兽药企业监控兽药制剂的质量,以及管理部门加强兽药质量监控,震慑不法掺假分子提供了技术保障,从源头上保证了动物源性食品的安全。
猜你喜欢
今日畜牧兽医(2022年10期)2022-12-23
核化学与放射化学(2022年2期)2022-04-28
化工自动化及仪表(2021年3期)2021-06-04
食品工程(2020年4期)2021-01-20
湖南农业(2020年7期)2020-09-16
中国油脂(2020年3期)2020-04-10
酿酒科技(2019年10期)2019-11-12
钻井液与完井液(2018年2期)2018-06-13
中国蜂业(2018年4期)2018-05-09
中南民族大学学报(自然科学版)(2015年2期)2015-12-16
