
迷迭香酸磁性分子印迹聚合物的合成及应用
2023-01-18 02:14:34韩祎陟罗云敬邹明强
分析科学学报 2022年6期
张 伟, 韩祎陟, 罗云敬*, 邹明强
(1.北京工业大学环境与生命学部,环境与病毒肿瘤学北京市重点实验室,北京100124;2.中国检验检疫科学研究院,北京 100123;3.呼和浩特国际旅行卫生保健中心,呼和浩特 010020)
迷迭香酸(Rosmarinic Acid,RosA)作为一种水溶性的天然酚酸类抗氧化剂,广泛存在于迷迭香、紫苏、薄荷等天然植物中,在保健品、食品和化妆品等领域中具有重要的实用价值。分离提取迷迭香酸的传统方法包括溶剂浸提法[5,6]、超临界萃取[7]、生物酶解[8 - 11]和大孔树脂吸附法[12,13]等。然而,这些方法的提取产物往往存在多酚类衍生物和蛋白等干扰物[5],特异性差且效率低。因此,开发一种具有选择性和实用性的迷迭香酸提取分离方法十分重要。
采用磁性分子印迹技术在铁、钴、镍等支撑材料表面合成分子印迹聚合物,生成磁性分子印迹聚合物(Magnetic Molecularly Imprinted Polymers,MMIPs)[11,14,15]。它同时兼具特异性和超顺磁性,在外加磁场的作用下,可以快速实现复杂基质中目标物的提取和分离,极大提高了传统分子印迹的吸附性能和分离效率[16,17]。目前,磁性分子印迹技术应用于食品安全[18,19]、药物分析[20,21]和环境保护[22]等领域。采用磁性分子印迹法提取分离迷迭香酸的研究未见报道,故本文以迷迭香酸为模板,丙烯酰胺(AM)为功能单体,偶氮二异丁腈(AIBN)为引发剂,乙二醇二甲基丙烯酸酯(EGDMA)为交联剂,以乙烯基修饰的Fe3O4纳米粒子为核,合成了迷迭香酸磁性分子印迹聚合物(R-MMIPs),对其结构和性能进行表征和吸附实验,结合高效液相色谱法实现对实际样品中迷迭香酸的分离和测定。
1 实验部分
1.1 仪器及试剂
高效液相色谱仪(Waters e2695 HPLC-DAD),红外光谱仪(Nicolet 6700,美国ThermoFisher),高分辨透射电子显微镜(Tecnai G2 F30,美国FEI公司),场发射扫描电子显微镜(S-4800,日立公司),振动样品磁强计(MPMS-3,美国Quantum Desig公司),脱色摇床(TS-A,晶玻实验仪器厂),顶置式搅拌器(TS-20,上海安谱科技股份有限公司),数控超声波清洗器(KQ-250DB,昆山市超声仪器有限公司)。
迷迭香酸(RosA)、丙烯酰胺(AM)、FeCl2·4H2O、FeCl3·6H2O、桑黄素(Morin)、辣椒碱(Capsaicin)、正硅酸乙酯(TEOS)、乙二醇二甲基丙烯酸酯(EGDMA)购于北京虹湖联合化工产品有限公司;甲基丙烯酸(MAA)、4-乙烯基吡啶(4-VP)、偶氮二异丁腈(AIBN)、硅烷偶联剂KH570(MPS)购于梯希爱(上海)化成工业发展有限公司;NH3·H2O(25%)、甲酸、冰乙酸、甲醇、乙醇购于福晨(天津)化学试剂有限公司。
1.2 迷迭香酸磁性分子印迹聚合物的制备
1.2.1 Fe3O4磁性纳米颗粒的制备使用化学共沉淀法制备Fe3O4纳米颗粒。将2.36 g FeCl3·6H2O和0.86 g FeCl2·4H2O溶于50 mL去离子水中,充分搅拌。随后缓慢加入5 mL NH3·H2O,70 ℃持续搅拌1 h。上述反应过程均在N2保护下进行。冷却至室温后用外磁场进行收集分离,用去离子水反复清洗至中性,50 ℃真空干燥24 h。
1.2.2 乙烯基修饰Fe3O4磁性纳米颗粒的制备采用溶胶-凝胶法在Fe3O4颗粒表面修饰SiO2。将500 mg Fe3O4纳米颗粒分散于100 mL乙醇/水混合液中,超声分散20 min。然后,在N2保护下依次加入8 mL NH3·H2O和4 mL TEOS。室温下搅拌反应8 h。通过外磁场分离产物,用乙醇和超纯水彻底清洗5次后,向瓶中加入100 mL乙醇,超声分散20 min。然后,在N2保护下缓慢加入6 mL MPS,40 ℃下搅拌反应12 h,反应产物经外磁场分离,并用乙醇和超纯水多次洗涤。最后,将产物在50°C真空干燥24 h。
1.2.3 迷迭香酸磁性分子印迹聚合物的制备将0.3 mmol迷迭香酸和1.8 mmol丙烯酰胺溶于40 mL乙醇中,置于4 ℃冰箱里静置12 h预聚合。然后加入200 mg乙烯基改性的Fe3O4@SiO2,在氮气保护下充分搅拌30 min。随后加入30 mmol EGDMA 和0.5 mmol AIBN,超声20 min后温置于60 ℃的搅拌器上反应24 h,全程N2保护。反应完成后,使用强磁铁分离聚合物,然后用甲醇∶乙酸(体积比9∶1)混合溶液索氏提取24 h,直至紫外分光光度计检测不到溶液中的迷迭香酸为止。最后用乙醇和纯水彻底清洗聚合物,并放入烘箱干燥。磁性非分子印迹聚合物(Magnetic Non-molecularly Imprinted Polymers,MNIPs)的制备与 MMIPs相同,但不加入迷迭香酸。
1.3 MMIPs和MNIPs的性能实验
1.3.1 MMIPs和MNIPs的吸附容量实验分别称取15 mg MMIPs和MNIPs置于5 mL梯度浓度(0~30 mg/L)的迷迭香酸乙醇溶液中。在室温下,振荡40 min后,用强磁铁分离聚合物。用紫外分光光度仪测定上清液中迷迭香酸的浓度。分别计算MMIP和MNIP对迷迭香酸的吸附容量Q,并绘制静态吸附等温线。
分别称取15 mg MMIPs和MNIPs置于5 mL迷迭香酸乙醇溶液(24 mg/L)中。室温下,振荡不同时间(0~80 min)。然后用强磁铁分离聚合物。用紫外分光光度仪测定上清液中迷迭香酸的浓度。分别计算MMIP和MNIP对迷迭香酸的吸附容量Q,并绘制吸附动力学曲线。
1.3.2 MMIPs和MNIPs的选择性实验选取迷迭香酸及其结构类似物桑黄素、辣椒碱用于选择性实验,通过比较聚合物对3种物质的吸附容量来对比其选择性。将15 mg MMIPs和MNIPs分别加入到盛有5 mL(30 mg/L)混合溶液的试管中。在室温下振荡40 min后,用强磁铁分离聚合物。取上清液用旋转蒸发仪浓缩,用3 mL甲醇复溶待测。结合HPLC法测定浓缩液中迷迭香酸和结构类似物的含量。按照上述方法测定MMIPs、MNIPs对不同物质的吸附容量,计算出分离因子(α)和印迹印子(β),考察两种聚合物的吸附选择性。
1.4 MMIPs的稳定性实验
准确量取5 mL迷迭香酸标准溶液(30 mg/L)加入到盛有25 mg MMIP或MNIPs的试管中。室温下,振荡40 min后用强磁铁分离聚合物。结合HPLC-DAD测定上清液中的迷迭香酸含量,同时用甲醇∶乙酸混合溶液、纯水清洗聚合物。反复循环10次,评价其稳定性。
1.5 色谱条件
色谱柱:Waters Sunfire C18柱(150 mm×4.6 mm,5 μm);流动相A:甲醇,B:质量分数0.1%的甲酸水溶液;等度洗脱:比例A∶B=60∶40;柱温:25 ℃;流速:1.0 mL/min;进样量:10 μL;检测波长:280 nm和330 nm。
1.6 MMIPs在实际样品中的应用
从当地农贸市场购买紫苏和紫甘蓝。分别准确称量10 g样品,充分干燥后破碎,加入60 mL 95%乙醇。300 W超声提取15 min,随后在70 ℃下回流提取2 h,过滤去渣,滤液通过旋转蒸发仪浓缩,用乙醇复溶至25 mL备用[23 - 25]。将30 mg MMIPs和MNIPs加入到5 mL上述提取溶液中,在室温下在振荡40 min后,使用磁铁分离,吸取3 mL上清液待测。将标液和上清液分别通过旋转蒸发仪浓缩,并使用甲醇复溶。经0.22 μm滤膜过滤后HPLC-DAD测定,重复3次,计算平均检出量、平均加标回收率和相对标准偏差(RSD)。
2 结果与讨论
2.1 不同功能单体对聚合物吸附容量的影响
在相同实验条件下,分别选取丙烯酰胺、甲基丙烯酸、4-乙烯基吡啶按照1.2.3节合成普通分子印迹聚合物,只是不加入磁性材料,分别考察功能单体种类和比例对聚合物吸附容量的影响。其结果如表1所示,以甲基丙烯酸为功能单体合成的聚合物吸附量明显低于其他聚合物,这可能是由于碱性功能单体与迷迭香酸更易形成稳定复合物,从而生成更多识别位点。在1∶4、1∶6和1∶8三个水平下,分别合成聚合物,可以看出,当模板分子与功能单体比例为1∶6时,该聚合物的吸附容量高于另外两个水平。因此选择丙烯酰胺为功能单体,与模板分子摩尔比为1∶6时合成聚合物。

表1 功能单体对吸附容量(Q)的影响
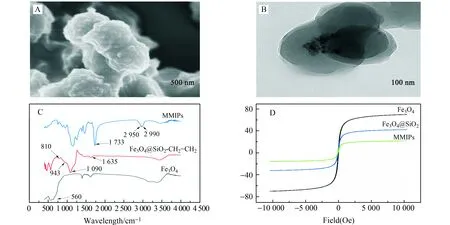
图1 MMIPs的SEM(A)、TEM(B)、红外光谱图(C)和磁滞曲线(D)Fig.1 SEM(A),TEM(B),infrared spectra(C)and hysteresis regression lines(D) of MMIPs
2.2 表征
2.2.1 MMIPs电镜表征图1A和B分别为MMIPs的扫描电镜(SEM)和透射电镜(TEM)图。SEM图中MMIPs纳米粒子表面粗糙,增加了聚合物的比表面积,有利于实现吸附和解吸附,其尺寸均匀,粒径约为200 nm。如TEM图所示,在硅球外侧观察到了颜色更浅的印迹层,形态均匀,证明了分子印迹层被涂覆在磁性支撑材料表面。
2.2.2 MMIPs和MNIPs的红外表征Fe3O4、Fe3O4@SiO2-CH=CH2和MMIPs的红外光谱如图1C所示。从Fe3O4的红外谱图可见,在560 cm-1附近为Fe-O的伸缩振动吸收峰。从Fe3O4@SiO2-CH=CH2的谱图可以看出,Si-O、Si-OH、Si-O-Si、C=C的伸缩振动峰分别出现在810、943、1 090和1 635 cm-1附近,这表明乙烯基成功地修饰到Fe3O4@SiO2表面。MMIPs的红外谱图中,1 733 cm-1、2 950 cm-1、2 990 cm-1处吸收峰分别与C=O伸缩振动、亚甲基的C-H伸缩振动、甲基的伸缩振动有关,表明EGDMA和AM已经聚合,成功制备了MMIPs。
2.2.3 MMIPs和MNIPs的磁性表征使用VSM考察Fe3O4、Fe3O4@SiO2和MMIPs的磁性能。磁滞回线如图1D所示,三者的饱和磁化强度分别为63.46、41.82和25.42 emug-1。三条曲线均关于原点对称,表明没有磁滞现象,证明Fe3O4、Fe3O4@SiO2和MMIPs具有超顺磁性。MMIPs的饱和磁化率可以实现外加磁场下的快速分离。
2.3 MMIPs与MNIPs的吸附性能试验
2.3.1 静态吸附实验图2A是MMIPs的吸附等温线。结果表明,当浓度小于15 mg/L时,MMIPs吸附容量随浓度的增加而快速增加。当初始浓度接近15 mg/L时,吸附曲线逐渐平稳,吸附量逐渐饱和。MMIPs的饱和吸附容量为8.68 mg/g,约为MNIPs(1.43 mg/g)的6.1倍。在不同浓度下,MMIPs对迷迭香酸的吸附效果均显著高于MNIPs,证明MMIPs对迷迭香酸具有良好的吸附效果。
2.3.2 动态吸附试验图2B是24 mg/L迷迭香酸溶液下,MMIPs和MNIPs的吸附动力学曲线,两种聚合物的吸附量随接振荡时间的增加而增加,但吸附速率逐渐降低,这表明聚合物上的结合位点逐渐吸附饱和。在40 min左右基本达到吸附平衡,其平衡吸附量为8.35 mg/g。然而在MNIPs中,吸附主要发生在MNIPs表面暴露的非印迹位点上,使得其更快地达到吸附平衡。该快速吸附为MMIPs的实际应用奠定了基础。

图2 MMIPs和MNIPs对迷迭香酸的吸附等温线(A)和吸附动力学曲线(B)Fig.2 Adsorption isotherms(A) and adsorption kinetic curves(B) of RosA by MMIPs and MNIPs
2.4 选择性实验
MMIPs和MNIPs对迷迭香酸、桑黄素和辣椒碱的吸附量如图3所示。结果表明,MMIPs对迷迭香酸的吸附量和特异性明显高于其他结构类似物。迷迭香酸、桑黄素和辣椒碱的印迹因子分别为4.98、2.65和1.37。桑黄素和辣椒碱的选择性系数分别为2.03和3.16。因为结构类似物在形状、大小和空间结构上与吸附位点不完全匹配,导致其吸附量低。这表明MMIPs能够特异性识别迷迭香酸。
2.5 稳定性试验
为了评价R-MMIPs的稳定性,在相同的实验条件下,测定了MMIPs对迷迭香酸连续吸附-解吸的吸附能力。如图4所示,MMIPs对迷迭香酸的吸附效率经6次循环后仅下降5.8%,印迹聚合物的三维孔隙非常稳定,洗脱过程影响很小。结果表明,MMIPs重复使用后对迷迭香酸仍具有较好的结合能力。

图3 MMIPs和MNIPs对迷迭香酸、桑黄素、辣椒碱的吸附容量Fig.3 Adsorption capacity of MMIPs and MNIPs for RosA,Morin and Capsaicin
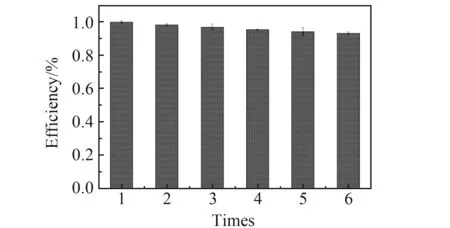
图4 MMIPs的重复性实验Fig.4 Repeatability test of MMIPs
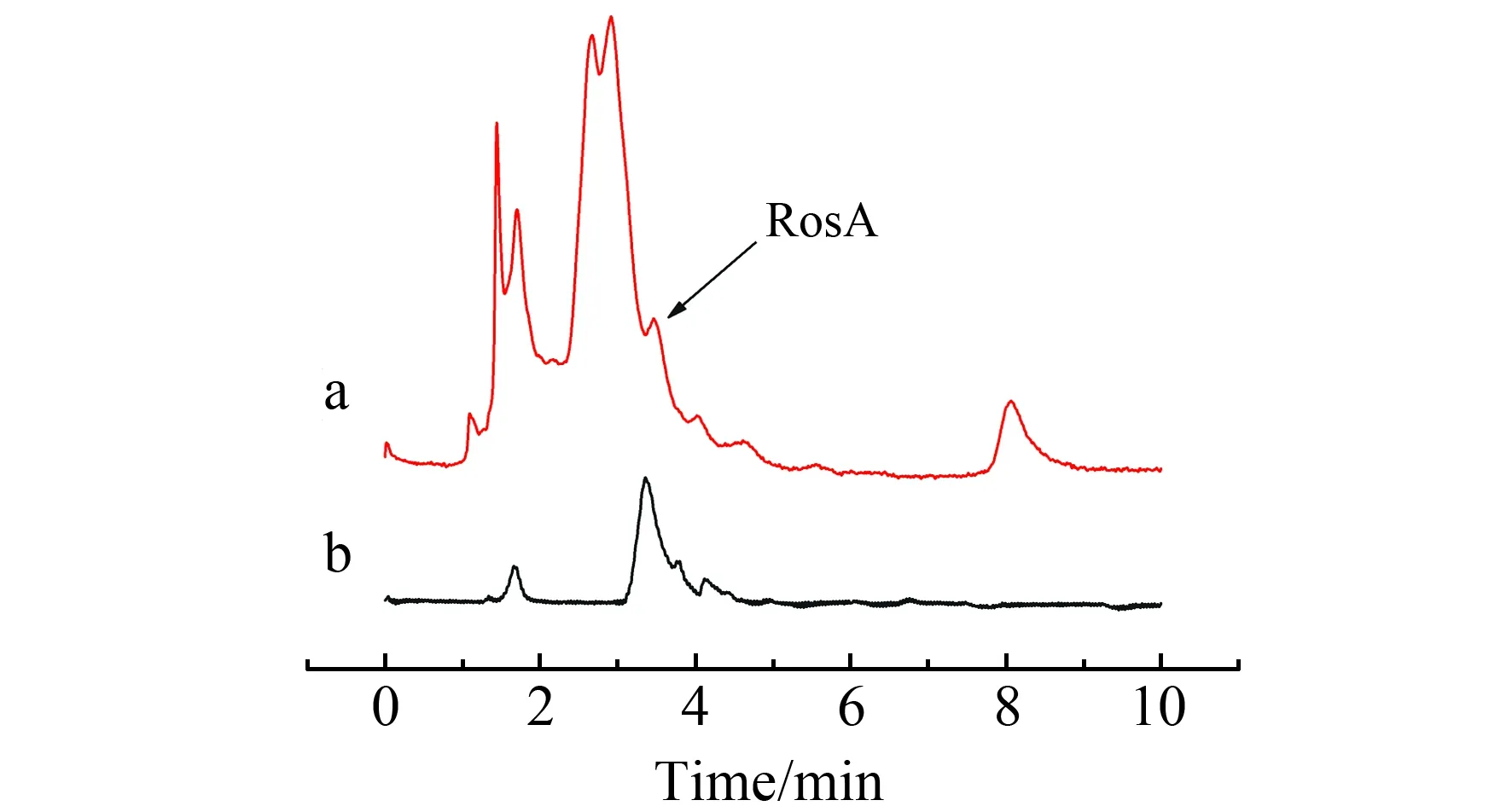
图5 加标样品(a)和R-MMIPs纯化后加标样品(b)色谱图Fig.5 Chromatograms of spiked sample(a) and spiked enriched sample(b)
2.6 实际样品中的应用
利用R-MMIPs对紫苏加标样品中的迷迭香酸进行分离和富集。如图5a所示,紫苏样品的提取液是一个复杂的基质。加标溶液经R-MMIPs富集后,溶液中干扰物含量明显减少(图5b)。结果表明,R-MMIPs对迷迭香酸具有明显的特异性识别和纯化能力。
方法学验证采用HPLC-DAD结合线性范围、相关系数、检出限(LOD)和定量限(LOQ)来考察。迷迭香酸在0.3~50 mg/L范围内线性关系良好,相关系数为0.9993。按信噪比的3倍和10倍计算,检出限和定量限分别为0.35 μg/mL和1.10 μg/mL。在15 μg/mL、25 μg/mL、35 μg/mL三个不同加标水平下向紫苏样品中添加迷迭香酸,对该方法的准确度和精密度进行评价(表2),加标回收率为88.1%~107.5%,相对标准偏差(RSD)为1.58%~4.42%(<5%)。结果表明,所建立的方法准确、可行、选择性好,可用于实际样品中迷迭香酸的提取和测定。
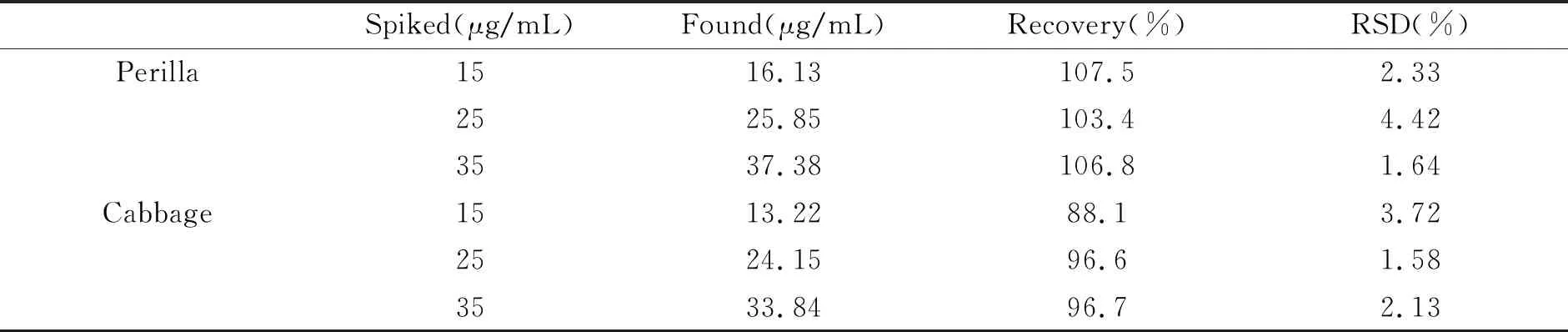
表2 样品中迷迭香酸的加标回收率(n=3)
3 结论
本文成功合成了迷迭香酸磁性分子印聚合物(R-MMIPs),其具有吸附能力强(Qmax=8.68 mg/g)、特异性高(印迹因子为4.98)和稳定性好等优点。对R-MMIPs微观结构进行了系统表征,该纳米材料粒径均匀,磁性强,在外加磁场下能够实现快速分离,无需离心或过滤。将MMIPs-HPLC成功应用于紫苏中迷迭香酸的分离和检测,样品加标回收率为88.1%~107.8%。为迷迭香酸等小分子天然产物的分离检测提供了一种高效的方法。
猜你喜欢
陶瓷研究(2022年3期)2022-08-19 07:15:18
云南画报(2021年10期)2021-11-24 01:06:56
纤维复合材料(2018年2期)2018-12-07 00:41:24
小学生优秀作文(高年级)(2018年4期)2018-09-11 01:23:22
军事文摘·科学少年(2017年4期)2017-06-20 23:22:10
材料科学与工程学报(2016年2期)2017-01-15 13:34:42
石油化工建设(2015年4期)2015-12-01 04:17:09
橡胶工业(2015年6期)2015-07-29 09:20:38
警察技术(2015年4期)2015-02-27 15:37:51
中国摄影(2014年12期)2015-01-27 13:57:04
