
根际细菌群落结构与装配对烟草根系生长性状的影响
2023-01-11 08:06:06侯建林李宏光肖艳松职齐琦许淘莎于法辉靳志丽周向平
激光生物学报 2022年6期
侯建林,李宏光*,肖艳松,职齐琦 ,胡 琦,许淘莎,于法辉,靳志丽,周向平
(1.湖南省烟草公司郴州市公司,郴州 423000;2.中南大学资源加工与生物工程学院,长沙 410083;3.湖南省烟草公司永州市公司,永州 425000)
植物根际微生物具有高度多样性[1],在生物地球化学循环中扮演了关键的生态角色[2]。根际微生物的群落组成和功能结构与能量流动和碳氮元素循环密切相关,部分功能微生物还可分泌植物生长因子,促进植物生长。例如,根际中存在多种可分泌植物生长素吲哚乙酸(indole-3-acetic acid,IAA)的微生物,包括根瘤菌(Rhizobium)[3]、芽孢杆菌(Bacillus)和乳酸杆菌(Lactobacillus)[4]等。此外,还有大量微生物可溶解根际矿物质并释放营养元素,如锌[5-6]、磷[7-9]、钾[10-11]等。然而,目前大部分关于促进植物生长的微生物研究主要聚焦于单菌的代谢功能上,而微生物间协同作用的研究较少。解析根际微生物功能团及其协同作用机制,不但可深入剖析根际微生物在根际中扮演的生态角色,还可为开发微生物菌肥以促进植物生长发育提供理论依据。
根际环境总体上塑造了微生物的群落组成[12]、多样性[13]、相互作用和发育装配模式[14]及功能结构[15-16],而微生物依其代谢活性营造了适宜生存的微环境[17-18],从而改善了植物的根际环境,提高了根系性状。一方面,土壤微生物可分泌胞外多聚物形成生物膜,以提高对环境扰动的抗性、群落多样性和代谢活性[18]。土壤微生物也会分泌铁载体,以促进环境铁离子的富集,提高微生物对重金属的抗性[19],强化植物对铁离子的吸收效率[20]。这些结果表明,微生物可以依靠其代谢功能显著影响土壤环境。另一方面,土壤微生物群落的分子生态网络会随着植物生长逐渐变得复杂,模块性及模块数量升高[21],这与构建氮降解等微生物功能团的形成密切相关[22],暗示微生物可在短期内快速参与根际土壤环境的形成。尽管大量研究表明了微生物群落结构特征在植物根系性状的改变中发挥着关键作用,强调了作物生长过程中微生物种群组成与根系性状差异的关联性,但目前对根际微生物的组成、共现网络、关键功能物种和发育装配及其对根系特征演变的作用仍知之甚少。
本文以发达根系(well-developed root system,WDR)与不发达根系(undeveloped root system,UDR)的根际系统为研究对象,利用16S rRNA高通量测序技术,研究农田根际微生物的群落组成,探究根系性状的差异与微生物群落组成的多样性、结构功能、分子生态网络拓扑性质及发育装配模式的关联性,用于解决以下问题:1)根际细菌群落在WDR与UDR的条件下是否存在显著不同?2)是否存在特别重要的微生物类群在装配过程中发挥关键作用?
1 材料与方法
1.1 试验设计
样本采集自湖南一块长期定位的试验烟田(经度 :112°06' N,纬度 :27°59' E),分别采集成熟期根系WDR和UDR的烟草(Nicotiana tabacum)根际土壤,每类各10份。采集过程中将整株植物连根拔起,去除根系松散附着的土壤,用刷子刷取根部黏附的剩余土壤即为根际土壤,标记保存后用于后续细菌群落分析和高通量测序分析。根际土壤的理化特性如表1所示,有效钾含量的平均值为787.58 mg/kg,有效磷含量的平均值为81.68 mg/kg,总氮量的平均值为2 039.20 mg/kg,有机质的含量百分比为4.19%,pH平均值为5.97。
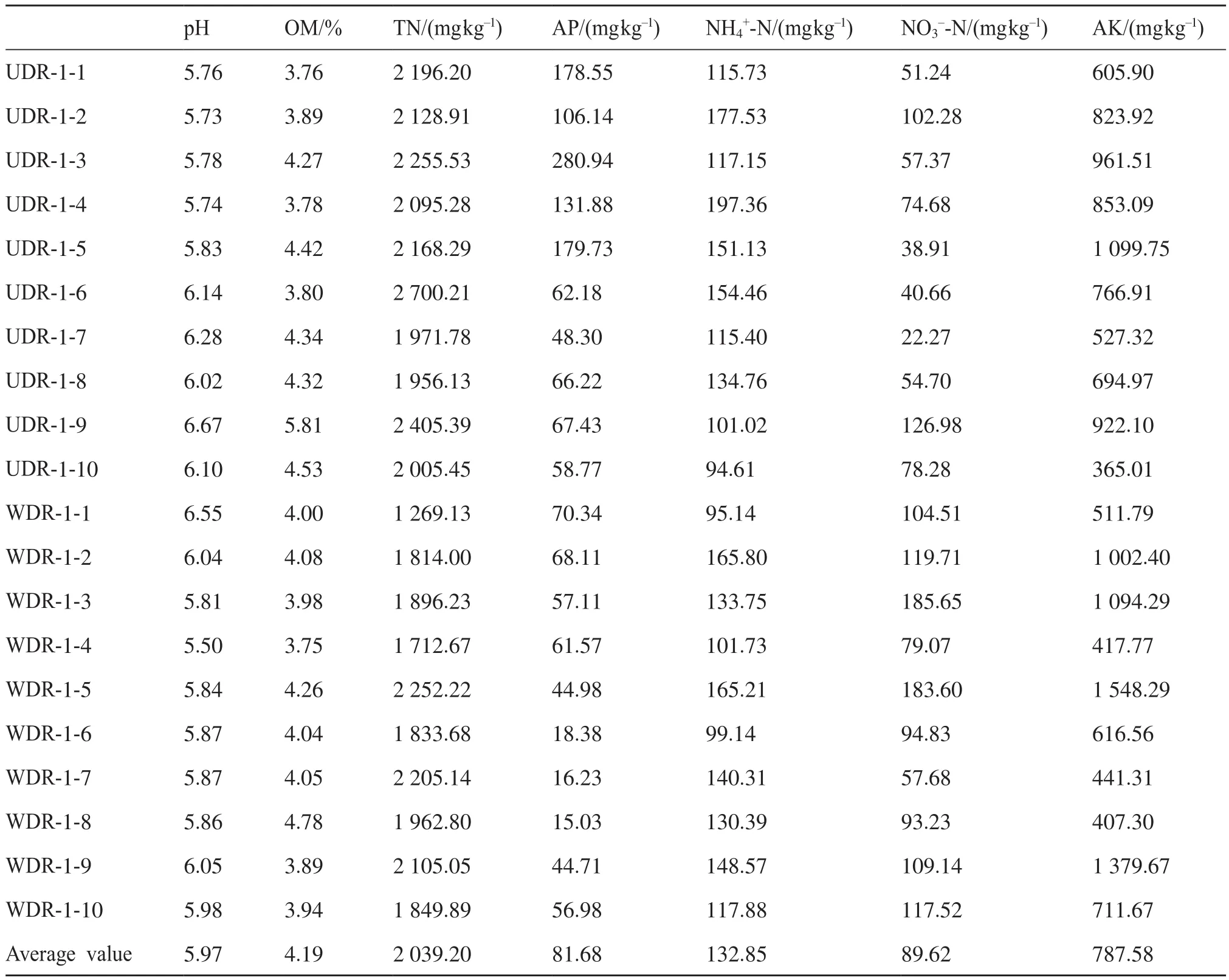
表1 根际土壤的理化特性分析Tab.1 Soil physicochemical properties
1.2 DNA提取和扩增子测序
利用强力根际DNA提取试剂盒(MoBio,SanDiego,CA,USA)按说明书从1.0 g根际样品中提取DNA。利用338F(5′-ACTCCTACGGGAGGCAGCA-3′)和 806R(5′-GGACTACHVGGGTWTCTAAT-3′)引物扩增细菌16S rRNA基因的V3~V4区。PCR扩增反应体系含有1.0 μL DNA模板(~25 ng DNA)、12.5 μL Taq Master Mix(Vazyme,Piscataway,NJ,USA)、0.5 μL引物,DNase-free去离子水调节体系体积至25.0 μL。PCR扩增程序设置如下:98℃ 30 s;98℃ 10 s,54℃ 30 s,72℃ 45 s,32次循环;72℃ 10 min;4℃保存。利用Gel Extraction Kit(OMEGA Bio-Tek Inc.,Doraville,GA,United States)纯化PCR产物。利用Qubit 2.0 Fluorometer(Invitrogen,USA)对其定量,等摩尔浓度混库后,在Illumina NovaSeq PE250平台上进行高通量测序(LC-Bio Technology Co.,Ltd,Hangzhou,Zhejiang Province,China),得到已去接头FASTQ格式的原始数据。
1.3 数据处理与分析
使用QIIME 2来处理原始数据,并且去除低质量序列和嵌合体,再生成操作分类单元(operational taxonomic unit,OTU)表,进行物种注释。使用R语言进行所有的计算分析,得到微生物群落Shannon、Pielou等多样性指数,采用主坐标分析(principal coordinates analysis,PCoA)来比较总体微生物群落结构。采用多重比较方差分析方法计算组间细菌群落的Alpha多样性差异。利用线性判别分析(linear discriminant analysis effect size,LEfSe)确定组间各分类水平上具有统计学差异的生物标识。基于Spearman相关性构建分子生态网络。利用零模型分析细菌群落系统发育装配。
2 结果与分析
2.1 根际细菌的群落组成和结构
利用16S rRNA基因高通量测序技术分别对UDR和WDR的根际细菌群落进行检测注释,结果如图1所示。在门水平上,UDR和WDR根际细菌群落的优势菌均主要为Proteobacteria、Acido-bacteriota、Bacteroidota、Chloroflexi和Patescibacteria,UDR根际细菌群落主要包含26.2%~46.3%的Proteobacteria、14.4%~26.6%的Acidobacteriota、2.6%~16.8%的Bacteroidota、2.1%~10.0%的Chloroflexi和2.1%~13.6%的Patescibacteria,而WDR根际细菌群落主要包含20.9%~32.5%的Proteobacteria、23.6%~34.4%的Acidobacteriota、3.0%~12.0%的Bacteroidota、5.9%~10.3%的Chloroflexi和2.6%~4.6%的Patescibacteria。
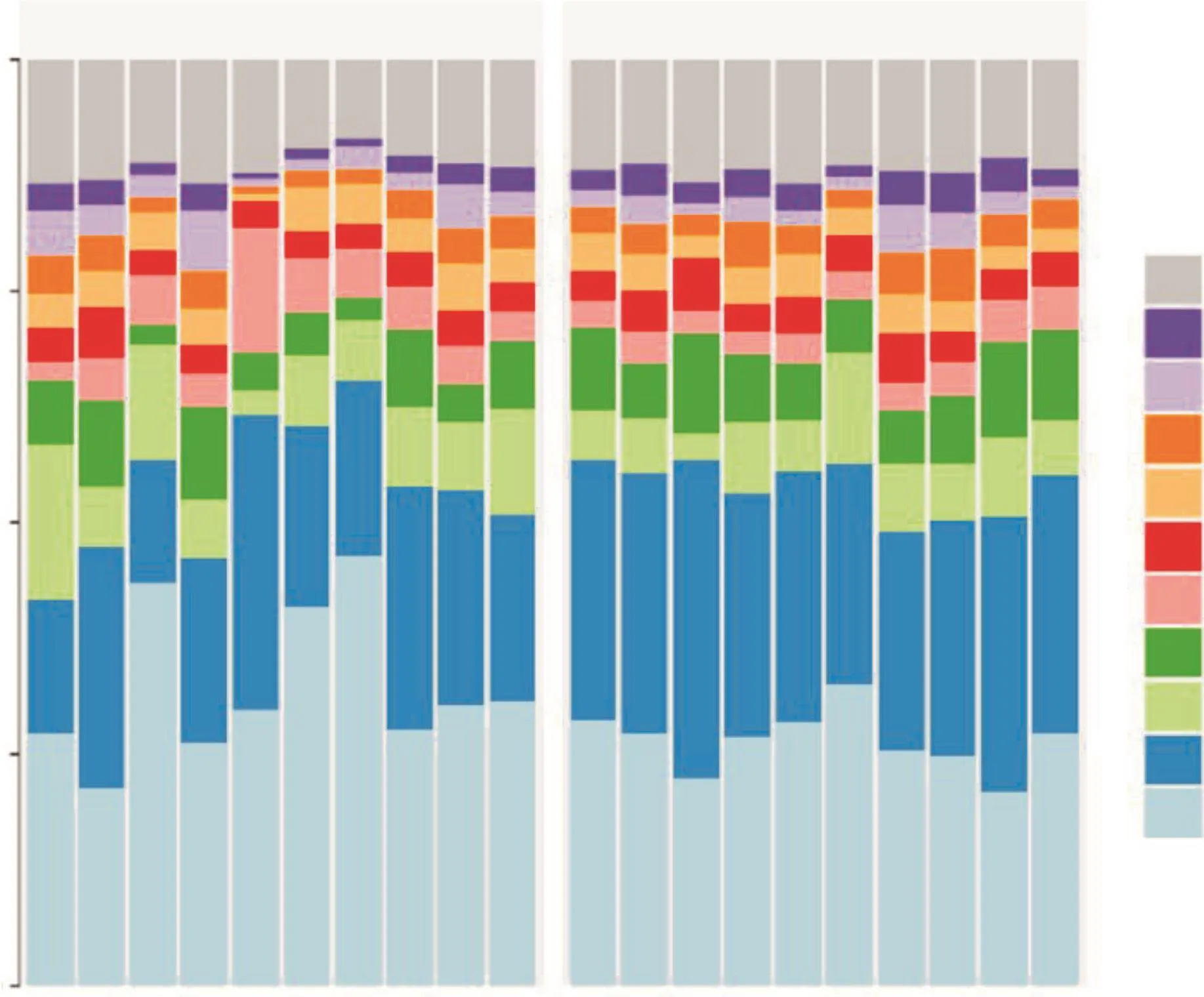
图1 UDR和WDR根际细菌群落的组成Fig.1 Rhizosphere bacterial community composition of UDR and WDR
Alpha多样性分析结果显示,WDR根际细菌群落的Observed、Chao1、ACE、Shannon、Simpson和InvSimpson指数均高于UDR,表明WDR根际细菌群落物种的丰度和多样性均高于UDR(表2)。
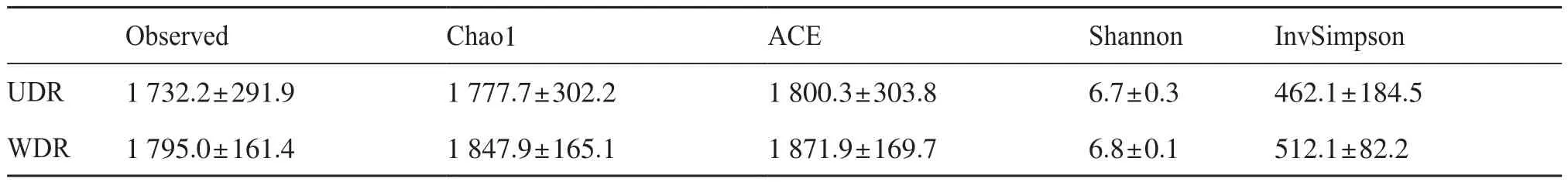
表2 UDR和WDR根际细菌群落的Alpha多样性分析Tab.2 Alpha diversity analysis of rhizosphere bacterial communities of UDR and WDR
PCoA结果显示(图2),WDR和UDR样本间明显分开,主坐标轴PCoA1和PCoA2分别解释了18.0%和14.2%细菌群落结构的差异,解释度较低,表明根际细菌群落结构是复杂的。
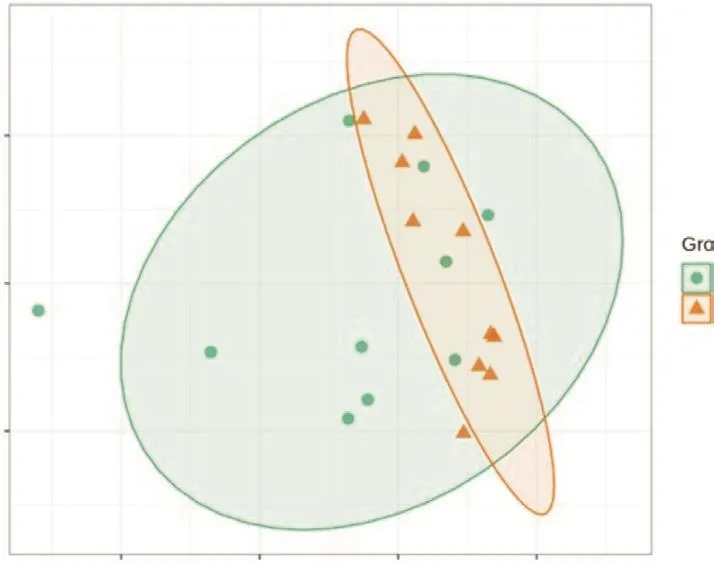
图2 UDR和WDR根际细菌群落相似性分析Fig.2 Rhizosphere community similarity analysis of UDR and WDR
通过差异性检验确定了WDR和UDR之间丰度具有显著差异的物种(P<0.05)。图3展示了丰度最高的16个差异物种,其中WDR根际细菌群落的Edaphobaculum、PLTA13、Rhodobacter、Parablastomonas、Noviherbaspirillum、JG36-GS-52、Algoriphagus和Rhodoplanes等属均显著低于UDR,而Gallionella和Luteimona等属则显著高于UDR。
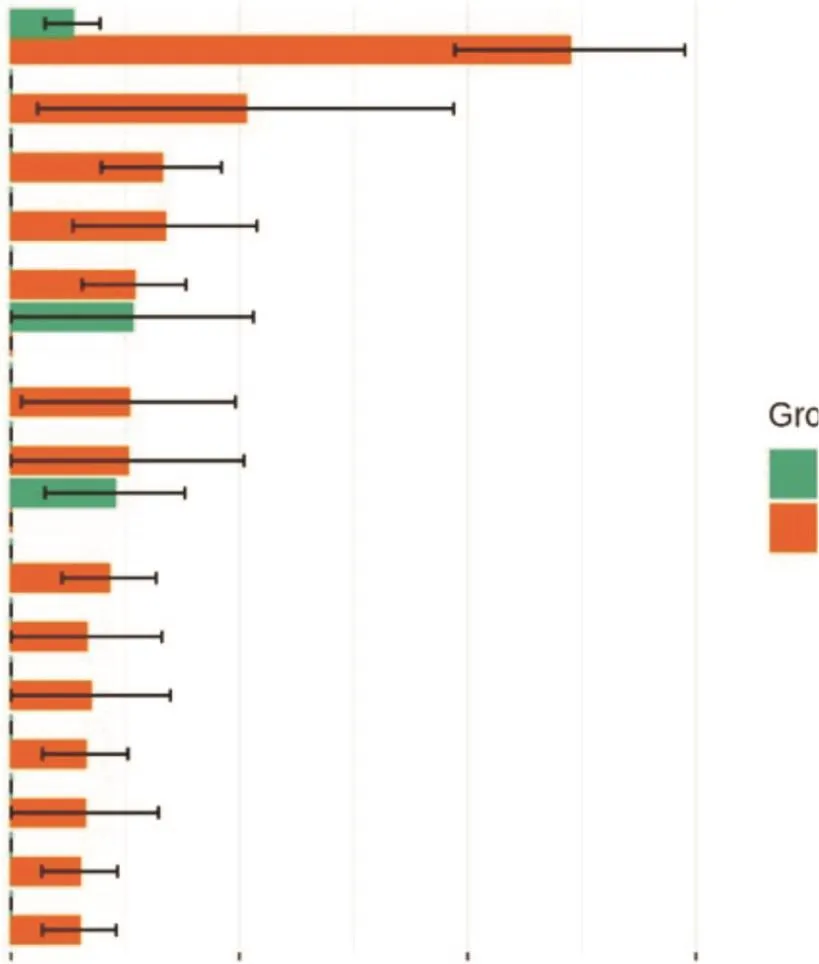
图3 UDR和WDR根际细菌群落差异性分析Fig.3 Difference analysis of rhizosphere bacterial community between UDR and WDR
2.2 根际细菌群落的分子生态网络结构和关键微生物
为解析土壤菌群物种间的共存模式,本研究基于随机矩阵理论构建了UDR和WDR的根际细菌群落的分子生态网络。如表3所示,相比WDR,UDR根际细菌群落的分子生态网络的规模(节点,vertex)、连通性(边,edge)、平均度(average degree)、聚类系数(clustering coefficient)、密度(density)和内聚性(centralization)均增加,表明UDR的分子生态网络更加复杂;而平均路径长度(average path length)和网络直径(network diameter)减短、网络正相关连接(positive links)的百分比增加,表明UDR物种间的相互作用增强,特别是合作行为。
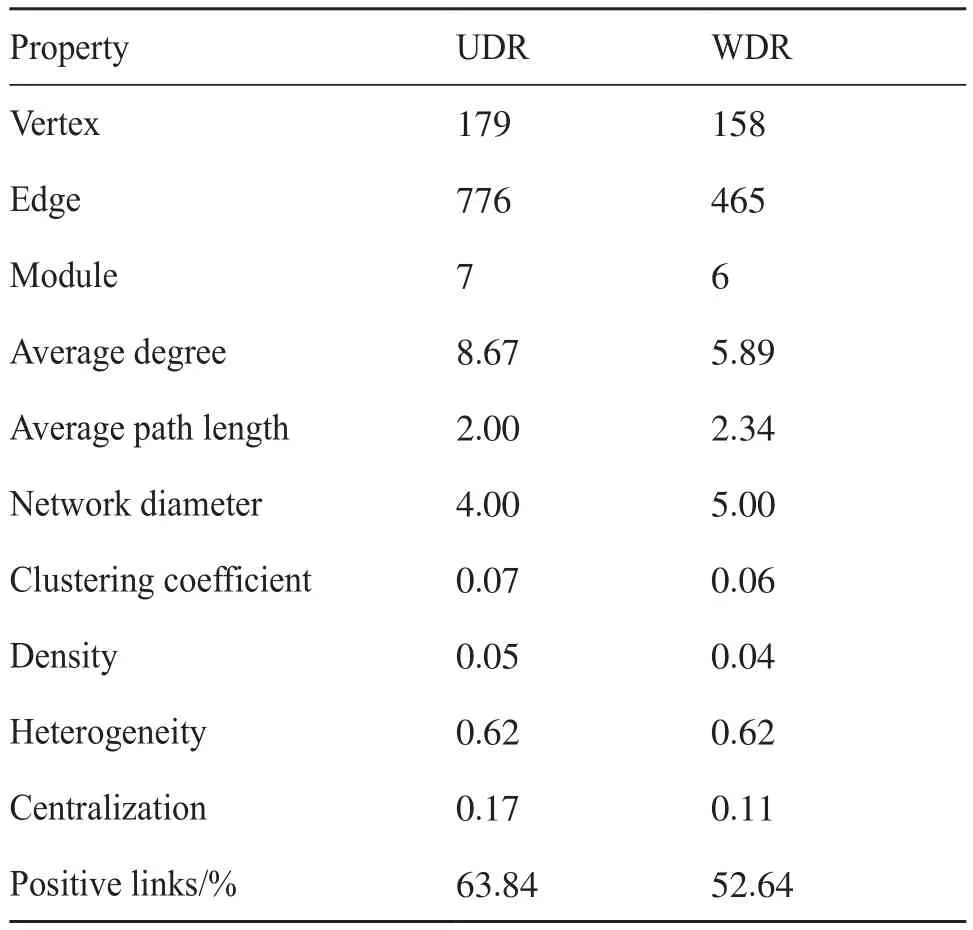
表3 UDR和WDR的根际细菌群落的分子生态网络拓扑性质Tab.3 Topological properties of molecular ecological network of UDR and WDR rhizosphere bacterial communities
网络结构的改变可能会进一步影响网络成员角色发生改变。基于它们的模块内连通度(Zi)和模块间连通度(Pi),检测到UDR的分子生态网络具有1个模块中心点[(moudule hubs;Zi>2.5且Pi<0.62),主要为Nitrospirota]、43个连接节点[(connectors;Zi<2.5且Pi>0.62),主要为Acidobacteriota和Proteobacteria]以及3个网络中心点[(Network hubs;Zi>2.5且Pi>0.62),主要为Proteobacteria和Gemmatimonadota];而WDR分子生态网络有2个模块中心点(主要为Acidobacteriota和Chloroflexi),52个连接节点(主要为Acidobacteriota和Proteobacteria)以及3个网络中心点(主要为Acidobacteriota、Nitrospirota和Proteobacteria)。这些都是在塑造网络结构中发挥关键作用的节点。相比UDR,WDR的关键节点数较多,且连接节点均主要属于Acidobacteriota和Proteobacteria。
网络模块与细菌群落功能的关联性预测结果显示,UDR和WDR的网络模块主要与能量流动功能相关。相比UDR,WDR的碳循环相关网络模块增加(图4)。

图4 UDR和WRD根际细菌群落网络模块的生物地球化学作用Fig.4 Biogeochemical effects of rhizosphere bacterial community network modules in UDR and WRD
2.3 根际细菌群落的发育装配
为了研究群落系统发育组装过程中确定性和随机性作用对根系发育情况的影响,本研究计算了净谱系亲缘关系指数(net related index,NRI)和最近种间亲缘关系指数(nearest taxon index,NTI)。相比UDR,WDR的NRI和NTI指数较高,表明WDR根际微生物群落装配更趋向于发育聚集(图5)。
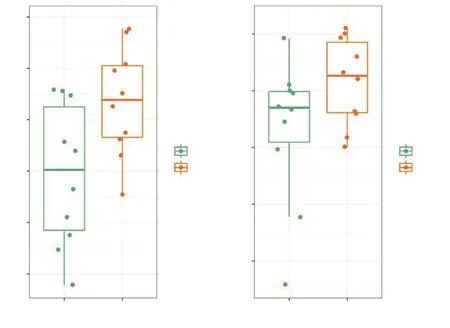
图5 UDR和WDR根际细菌群落NRI和NTI指数Fig.5 NRI and NTI of rhizosphere community in UDR and WDR
图6进一步表明,WDR和UDR的betaNRI和betaNTI指数存在显著差异(P<0.000 1),且WDR的betaNRI指数小于-2,显著大于UDR,表明环境过滤在WDR的群落装配中发挥了更重要的作用。WDR根际细菌群落装配更趋向于发育聚集,且环境过滤在其中发挥了更重要的作用。
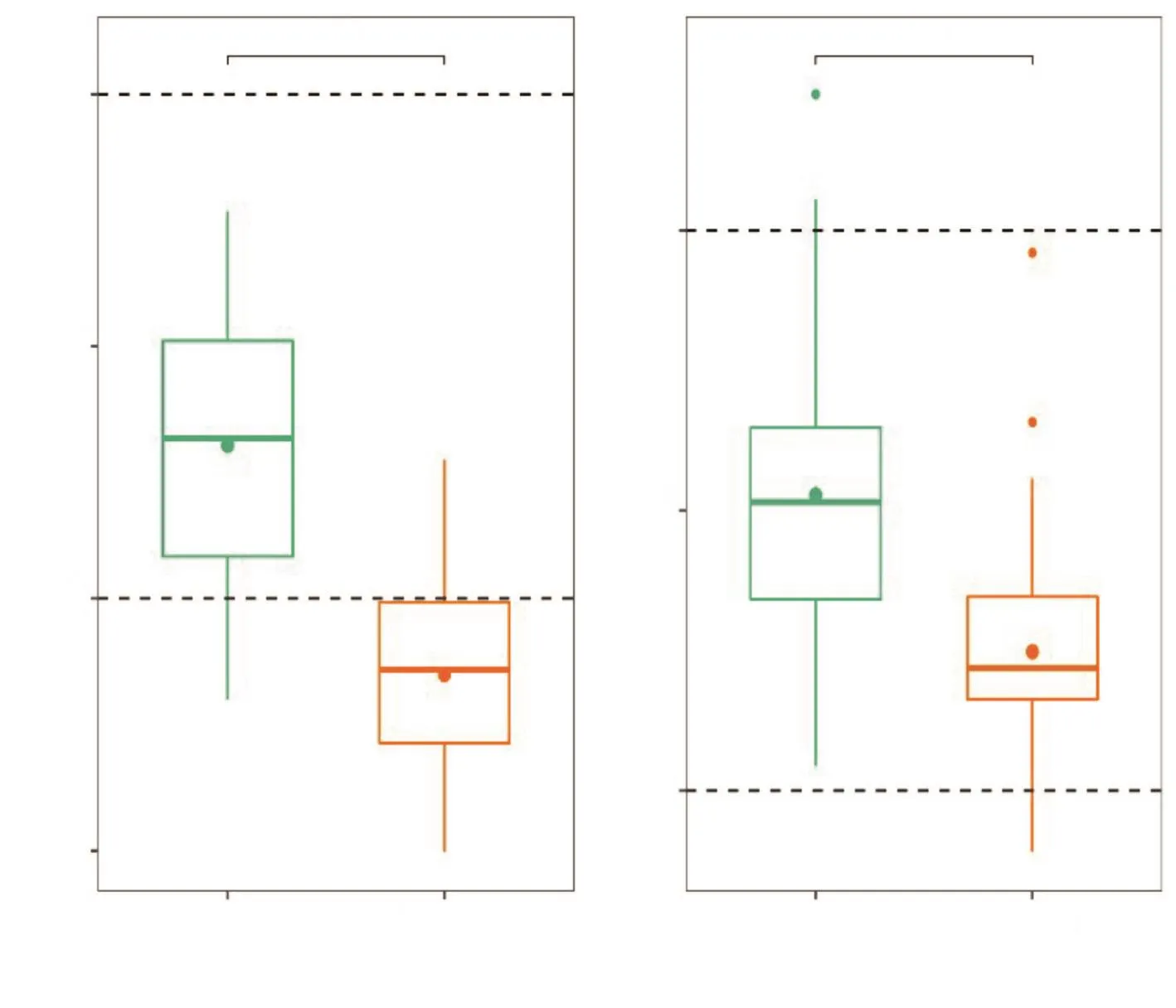
图6 UDR和WDR根际细菌群落betaNRI和betaNTI指数Fig.6 Rhizosphere community betaNRI and betaNTI index of UDR and WDR
3 讨论
根系微生物在烟草生长发育过程中有着重要的作用,其群落结构和发育装配与根系是否发达密切相关。Shi等[21]的研究结果表明,燕麦的根系发育过程与微生物群落密切相关,随着根系的发育,根际微生物的相互作用网络变得更加复杂和模块化;同时,随着根系的发育,根际微生物群落多样性降低,出现发育聚集的现象。本研究结果指出,WDR与UDR烟草的根际细菌群落组成存在显著差异。不同的是,WDR的根际细菌物种的丰富度和多样性均高于UDR,表明微生物群落结构对烟草根系发育产生了直接影响。与UDR相比,WDR的根际细菌群落中Gallionella和Luteimona等属的丰度显著较高,而Edaphobaculum、PLTA13和Rhodobacter等属的丰度则显著较低。Gallionella是一类铁氧化菌,是植物根系环境中Fe(II)的关键参与者。有研究指出,Gallionella相关的FeOB能参与并影响植物培育土壤中Fe(II)的氧化过程,促进根系铁元素的吸收[22]。而Luteimona是一类常见的植物促生菌,能通过影响植物根系的羧酸和胺的含量提高植物的抗逆生长能力[23]。
土壤微生物群落的相互作用是维持根际生态系统功能和结构稳定的关键[24],这些相互作用广泛且复杂。分子生态网络拓扑性质表明,相比WDR,UDR根际细菌群落的分子生态网络的规模、连通性、平均度、聚类系数、密度和内聚性增加,而平均路径长度和网络直径减短,表明不发达根系分子生态网络更加复杂,物种间相互作用更加强烈,且其网络正相关连接增加,表明UDR的根际细菌物种间相互趋向合作而不是竞争,具有更多正相互作用的复杂的分子生态网络,在环境变化中更有利于维持系统的稳定[25],但缺乏对有限资源的竞争以及独特的环境生态位和空间隔离,可能不利于根际环境的改善和植物的生长。模块特征基因分析对于揭示高阶组织和识别关键种群具有重要意义[26]。通过计算节点模块内连通度(Zi)和模块间连通度(Pi)检测得到WDR分子生态网络中的关键节点主要属于Chloroflexi和Proteobacteria。大量研究表明,Proteobacteria在微生物氮、磷循环中发挥着关键作用[27],Chloroflex的丰度对环境pH的变化较为敏感[28]。同时,相比UDR,WDR的碳循环相关网络模块增加。这些结果表明,网络模块化结构与微生物的特异性功能团形成有密切联系,深入剖析模块成员的组成及其相互作用将更有利于认识微生物群落在土壤物质循环的生态角色。
NRI可提供种群深度关联程度的重要信息,而NTI则能进行更精细的系统发育检查。结果表明,UDR和WDR的NRI/NTI值均大于0,表明两种情况下,微生物都呈现聚集。相比之下,后者的NRI/NTI指数大于前者,表明WDR的根际微生物发育更聚集。研究表明,植物与根系微生物的相互作用是其提高环境适应性和抗逆性力的重要因素,相反,环境因素也会影响微生物的群落组装[29]。可以初步判断,微生物聚集差异是导致植物根系发育差异的重要原因。然而,根系的发育情况和微生物群落装配的直接关系还有待进一步验证。
我们的研究结果表明了作物根系生长发育情况与微生物的群落组成、结构功能、网络变化和发育装配存在显著的关系。WDR根际系统的微生物群落的多样性高于UDR根际系统;WDR的根际系统Gallionella和Luteimona的丰度高于UDR的根际系统,推测其可能参与了根系的铁元素吸收和抗逆性的形成,从而促进了植物根系的生长。同时,UDR系统的分子生态网络较为复杂,物种间相互作用较强,特别是合作行为,而WDR系统的关键节点数和碳循环相关网络模块与UDR相比增加,网络结构变化似乎与微生物群落的功能结构和生态系统功能过程密切相关。此外,WDR的根际系统中,微生物群落系统发育更趋于发育聚集,表明微生物群落受环境选择的影响较大,暗示了根系生长过程中,微生物群落的系统发育是保守的。这些结果说明了微生物群落的关键物种可改善植物根系生长发育,为开发微生物菌肥调控根际微生物群落组装,从而促进植物根系生长提供了理论依据。
猜你喜欢
昆明医科大学学报(2022年2期)2022-03-29 00:51:58
中国土壤与肥料(2021年5期)2021-12-02 01:06:26
食品安全导刊(2021年20期)2021-08-30 06:40:50
四川劳动保障(2021年3期)2021-06-09 07:08:56
天然产物研究与开发(2018年3期)2018-05-07 06:38:35
现代园艺(2017年13期)2018-01-19 02:28:05
中国蔬菜(2016年8期)2017-01-15 14:23:38
西南农业学报(2016年5期)2016-05-17 05:42:36
中国农业文摘-农业工程(2016年5期)2016-04-12 05:38:09
水生生物学报(2015年1期)2015-02-28 16:01:05
