
一种噬菌体质粒的构建及其在抗体库模型筛选中的应用
2023-01-10 03:17:36康晓彤赵博
石河子大学学报(自然科学版) 2022年5期
康晓彤,赵博
(上海交通大学药学院,上海 200240)
1982年表达蛋白于病毒的表面被首次提出,Smith[1-2]于3年后将这一理念付诸实践,引出噬菌体展示技术(Phage display technology, PDT)这一概念,将外源基因表达的多肽展示在噬菌体的表面,后来Gregory P. Winter等利用该技术将抗体片段展示在噬菌体表面[3],使有关噬菌体领域迅速发展。
噬菌体展示技术是一种基于噬菌体表面展示的目标多肽或蛋白进行筛选的体外选择系统。其基本原理是将包含目的基因的质粒转入噬菌体内,表达在噬菌体的表面,根据特异性结合能力的强弱筛选出与靶分子亲和力高的目标多肽[4-6]。采用噬菌体展示技术,基因组允许插入外源DNA序列长度达12 000 bp[7]。噬菌体展示技术通常通过各种展示文库来筛选特异性探针[8]、研究抗原表位[9]、发现新型生物标志物[10]、生产高效低价疫苗[11]、研究DNA结合蛋白及蛋白质定向改造[12]等。本实验将编码整个抗体结合域的基因(以单链可变片段scFv形式)融合到基因III,以活性片段scFv形式表达,与完整抗体相比在组织穿透力方面都有明显优势,这种方法促进了在噬菌体病毒粒子外表面上足够水平的抗体展示,从而可选择识别抗原的噬菌体。
在本研究中提供了1种用于构建噬菌体抗体库的模型质粒,以及应用该质粒建立的模型筛选方法。在目的基因下游添加牛凝血酶(thrombin)特异性酶切位点,通过PCR引物扩增已有的抗BCMA(人源化的抗5666B细胞成熟抗原(B-cell maturation antigen))的单克隆抗体的scFv,将扩增的PCR片段克隆到pComb3H噬菌粒中,得到目标质粒pComb3H-BCMA-scFv。利用噬菌体表面展示BCMA-scFv建立噬菌体筛选模型,评估创新筛选方法的可行性,通过SDS-PAGE及Western Blot实验验证牛凝血酶特异性洗脱效果,利用ELISA(酶联免疫吸附实验)及噬菌体滴度检测特异性洗脱后单链抗体的结合活性。实验结果显示:在构建噬菌体抗体库的噬菌粒载体上添加牛凝血酶特异性酶切位点的筛选方法,可以减少筛选轮次高效筛选出特异性噬菌体抗体。
1 材料与方法
1.1 材料
供试菌株E.coliXL1-Blue,SS3220与噬菌粒载体pComb3H为本实验室储存,辅助噬菌粒M13KO7于Invitrogen公司购买。TaqDNA Polymerase于Toyobo公司购买,T4 DNA Ligase于Thermo Fisher Scientific公司购买,限制性内切酶SacⅠ、SpeⅠ、KpnI于New England Biolabs公司购买。离心柱型质粒小提试剂盒、通用型DNA回收试剂盒于天根公司购买,鼠源单抗anti-Flag、anti-M13于abcam公司购买,羊抗鼠单抗HRP goat-anti-mouse于CST公司购买,牛凝血酶Thrombin于翌盛生物科技有限公司购买,链霉亲和素96孔板于Thermo Fisher Scientific公司购买,TMB显色液于雷根公司购买,PCR引物合成、基因片段合成以及基因测序为GENEWIZ公司。TBS缓冲液(0.02 mol·L-1Tris HCl,0.15 mol·L-1NaCl,pH 约7.5),TBS-T缓冲液(0.05 mol·L-1Tris Base,0.15 mol·L-1NaCl,0.05%Tween,0.05%Triton X-100,pH约7.5)及其它试剂均在本实验室配置。
1.2 噬菌体抗体库模型质粒构建
1.2.1 以BCMA-scFv基因为模板进行PCR基因扩增
在BCMA-scFv(抗BCMA的单链抗体)基因后添加Flag小肽,作为后期Western Blot及ELISA验证的抗体标签,上游酶切位点SacI,下游SpeI酶切位点后添加牛凝血酶最适切割位点LVPRGS,设计抗体基因的上下游引物(表1)。以BCMA-scFv基因的质粒为模板,设计PCR体系为50 μL:引物浓度10 μmol·L-1,1 μL质粒,1 μL引物(上),1 μL引物(下),25 μLTaqMix,22 μL dd H2O。程序如下:94 ℃反应3 min预变形,94 ℃继续反应30 s,65 ℃反应30 s,72 ℃反应1 min,变形、退火、延伸过程进行30个循环,循环结束后72 ℃反应5 min,4 ℃暂存,得到BCMA-scFv的PCR产物。
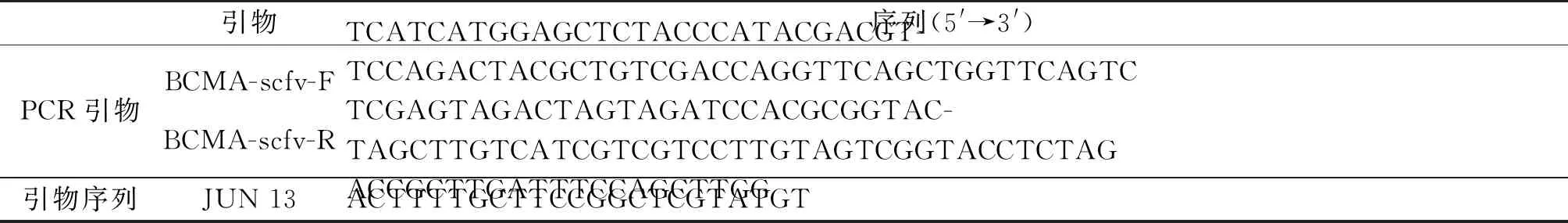
表1 PCR引物及测序引物信息
1.2.2 样品凝胶电泳
全部样品上样电泳(根据DNA分子量大小,配置1.2%琼脂糖凝胶),110 V,30 min。跑胶完成后,将凝胶置于凝胶成像仪中紫外下观察,拍照观察后,切取对应位置(约800 bp)的PCR片段,使用天根通用型DNA回收试剂盒回收纯化,使用微量分光光度计测定回收浓度。
1.2.3 连接转化测序单克隆
将BCMA-scFv基因进行SacⅠ和SpeⅠ酶切处理,连接在同样酶切位点的pComb3H载体上。30 μL酶切体系如下:800 ng BCMA-scFv PCR片段/1 μg pComb3H,1 μLSacI,1 μLSpeI,5 μL 10X buffer ,加dd H2O补齐至30 μL。将体系置于37 ℃温水中4 h,酶切结束后将产物进行琼脂糖凝胶电泳,浓度为1%,将跑完的凝胶置于紫外成像仪下从凝胶中切取对应位置的载体片段,使用天根通用型DNA回收试剂盒回收纯化。利用T4 DNA Ligase对Insert和Vector进行连接,设计连接体系为10 μL:7 μL BCMA-scFv酶切片段,1.5 μL pComb3H酶切片段,1 μL 10× T4 buffer,0.5 μL T4 ligase。混匀后室温反应1 h,将酶连产物3 μL通过化学转化的方式转化至DH5α细胞中,放于冰上30 min,42 ℃热激90秒,然后冰敷2 min,迅速加入SOC复苏培养基1 mL,放于37 ℃小摇床上培养1 h,将400 μL复苏液均匀涂布在含有氨苄抗生素的LB平板上,于37 ℃恒温箱中培养约12 h;第2天,从平板上挑取单克隆于摇菌小试管中接种,培养基为5 mL含有100 mg·mL-1氨苄抗生素的LB液体,于37 ℃小摇床220 rpm培养12 h;第3天,质粒小提试剂盒进行质粒抽提,于GENEWIZ公司进行测序。然后比对模板序列和测序结果,确定目标序列是否成功构建,得到质粒pComb3H-BCMA-scFv。
1.3 噬菌体表面展示BCMA-scFv
取2 μL测序正确的质粒电转化至被辅助噬菌体(helper phage)感染的50 μL SS320感受态细胞中,加1 mL预热SOC复苏,于37 ℃小摇床培养1 h,将全部复苏液移至2YT培养基,培养基包含氨苄和卡那双重抗性,放于37 ℃大摇床中250 r·min-1培养12 h;第2天,离心收集上清,5 000 r·min-110 min,于上清中加入20%的PEG-NaCl试剂,冰上静置至少30 min,经9 000 r·min-1离心20 min后,收集噬菌体沉淀,加入少量TBS溶液溶解沉淀,收集后4 ℃高速离心10 min,移上清于EP管中,弃细菌沉淀。取1 mL事先培养至OD600=0.6的XL1-Blue菌液,接种1 μL噬菌体溶液,37 ℃小摇床中250 r·min-1培养1 h,将10 μL菌液以10倍梯度进行稀释,在琼脂平板上点板,37 ℃恒温箱中培养约12 h,取出平板后计数菌落数来确定噬菌体滴度。
1.4 验证牛凝血酶洗脱效果
1.4.1 SDS-PAGE检测thrombin酶切噬菌体表面展示的BCMA-scFv
用酶切单位依次增加的牛凝血酶量0 U,2 U,4 U,6 U,8 U分别酶切100 μL表面展示BCMA-scFv的噬菌体,37 ℃水浴酶切2 h。酶切后,取40 μL样品加入10 μL 5×蛋白上样缓冲液,于金属浴99 ℃加热10 min使蛋白变性。根据目的蛋白及牛凝血酶蛋白大小,样品取40 μL使用8%~16%的梯度胶进行上样,以上层浓缩胶80 V,20 min,下层分离胶120 V,90 min进行SDS-PAGE电泳。电泳结束后,用考马斯亮蓝染料染色半小时,过夜脱色,隔日观看酶切蛋白情况。
1.4.2 Western Blot检测thrombin酶切噬菌体表面展示的BCMA-scFv
Western Blot实验前,酶单位依次增加的每种样品取35 μL上样,以90 V ,90 min的条件进行SDS-PAGE电泳,蛋白胶浓度为12%。电泳结束,将SDS-PAGE胶小心从玻璃板上切下,准备转膜。将夹子在盛有转膜液的水槽中打开,两侧的海绵垫上分别铺2层滤纸,在水中打湿,按照“黑胶白膜”的方式将SDS-PAGE胶和0.45 μm的NC膜贴合在滤纸上,转膜液没过胶和膜,保证NC膜可以完全覆盖胶上的Marker及样品条带,并用楔子赶走胶与膜之间的气泡。将夹子夹紧后随转膜液一起倒入电泳槽中,并将整个电泳槽插入冰中,保证转膜过程中的持续低温状态。调节电压100 V转膜80 min。转膜结束后,将NC膜浸入丽春红溶液中几分钟,清洗几次后观察膜上的条带状况,然后将丽春红洗净后将膜完全浸于5%脱脂牛奶中常温封闭1 h。封闭结束后,洗去NC膜上残留的牛奶,将膜置于含有Rabbit-HA一抗的塑封袋中,去除干净塑封袋中的气泡,4 ℃过夜孵育。隔日将NC膜置于1×TNET溶液中,洗膜3次,每次15 min。洗膜充分后,将膜置于含有R800荧光二抗的塑封袋中,避光孵育1 h。用上述方法洗去未结合的二抗,最后利用Odyssey双色红外荧光成像系统进行扫膜观察结果。
1.4.3 包被BCMA抗原通过ELISA和测试洗脱噬菌体滴度检测牛凝血酶洗脱效果
在链霉亲和素孔板中加入生物素化的BCMA抗原,于室温下孵育1 h,TBS溶液进行洗涤除去多余的抗原,用3%BSA/TBS溶液封闭未结合部位1.5 h,弃去封闭液后加入适量的噬菌体溶液,于37 ℃恒温箱中孵育1 h,TBS-T溶液洗涤去掉多余的噬菌体。在各实验孔中分别加入牛凝血酶单位依次增加0 U,2 U,4 U,6 U,8 U的洗脱液,TBS稀释至每孔100 μL,于37 ℃恒温箱中孵育2 h,收集与抗原结合的噬菌体,每100 μL洗脱液感染1 mL OD600=0.6的XL1-blue细胞,测试洗脱噬菌体滴度,同时将100倍稀释的鼠源anti-M13抗体加入洗脱后的孔板内,稀释液为TBS-T试剂,于37 ℃恒温箱中孵育1 h,TBS-T溶液洗涤除去多余的抗体,加入现用现配的TMB显色液,于室温下避光孵育15 min,用2 mol·L-1H2SO4溶液终止反应,酶标仪读取OD450数值。
1.4.4 包被anti-Flag抗体通过ELISA和测试洗脱噬菌体滴度检测牛凝血酶洗脱效果
在实验孔内加入1000倍稀释的鼠源anti-Flag抗体,稀释液为TBS试剂,室温下孵育1 h,TBS溶液洗涤除去多余的抗原,用3%BSA/TBS溶液封闭未结合部位1.5 h,弃去封闭液后加入适量稀释为1×、10×、100×的噬菌体溶液,稀释液为TBS-T试剂,37 ℃恒温箱孵育1 h,用TBS溶液和TBS-T溶液洗涤30次,尽可能除去未结合的噬菌体。在各实验孔中分别加入牛凝血酶单位依次增加0 U,2 U,4 U,6 U,8 U的洗脱液,TBS稀释至每孔100 μL,于37 ℃恒温箱中孵育2 h,收集与抗原结合的噬菌体,每100 μL洗脱液感染1 mL OD600=0.6的XL1-blue细胞,测试洗脱噬菌体滴度,同时thrombin洗脱后将100倍稀释的鼠源anti-M13抗体加入洗脱后的孔板内,稀释液为TBS-T试剂,于37 ℃恒温箱中孵育1 h,TBS-T溶液洗涤多余的抗体,加入现配的TMB显色液,于室温下避光孵育15 min,用2 mol·L-1H2SO4溶液终止反应,酶标仪读取OD450数值。
1.4.5 通过模型筛选分析牛凝血酶特异性洗脱效果
我们建立一种模型筛选方法,目的是从展示B7-H3-scFv(抗B7-H3的单链抗体,与BCMA无作用)的噬菌体混合物中富集展示BCMA-scFv的目标噬菌体,通过筛选轮次和目标噬菌体富集度评估筛选方法可行性。
1.4.5.1 B7-H3-scFv质粒构建
将B7-H3-scFv基因进行SacⅠ和KpnⅠ酶切处理,连接在同样酶切位点的pComb3H载体上,通过T4 DNA Ligase进行连接反应,经化学转化的方式转化至DH5α感受态中。其余与1.2中噬菌体抗体库模型质粒构建过程一致,保留正确的目的基因质粒。后挑选测序正确的质粒进行噬菌体制备。
1.4.5.2 模型筛选
在链霉亲和素孔板中加入300 μL BSA-TBS封闭液,封闭酶标孔中非特异性结合位点,室温下放置于水平摇床振摇2 h。弃去封闭液,在封闭处理过的孔板内加入生物素化的BCMA抗原,对照管中加入等量抗原包被液,于室温徐徐振摇1 h,TBS及TBS-T溶液先后洗涤酶标孔10次,以除净未结合的生物素化抗原。在展示B7-H3抗体的噬菌体溶液中分别混入稀释比例为1∶100,1∶1 000,1∶10 000的BCMA-scFv噬菌体,将噬菌体溶液混合后加入孔板内,结合1 h,先后用TBS溶液和TBS-T溶液洗涤30次,以除净多余的噬菌体。洗涤后,每孔加入100 μL 2 U酶切单位的牛凝血酶洗脱特异性结合噬菌体,37 ℃酶切2 h。将筛选组全部筛选后噬菌体溶液(洗脱液)合并,取1 mL事先培养至OD600=0.6的XL1-Blue菌液,接种1 μL噬菌体溶液,于37 ℃小摇床中250 r·min-1培养1 h,将10 μL菌液以10倍梯度进行稀释,在琼脂平板上进行点板,在37 ℃恒温箱中培养约12 h,取出平板后计数菌落数来确定第一轮筛选后能与抗原结合的噬菌体的滴度。
随着筛选的进行,可以通过减少链霉亲和素管内包被的抗原含量、减少抗原抗体结合时间以及降低噬菌体加入量等方式使得筛选条件更加严苛,达到寻找最优亲和力抗体的目的。根据每轮筛选富集度来判定是否需要继续筛选。
2 结果
2.1 构建新的质粒并通过SDS-PAGE和Western Blot检测噬菌体表面展示的BCMA-scFv
将双酶切处理的BCMA-scFv Insert产物连接至相同酶切位点的pComb3H载体上,对酶切产物进行琼脂糖凝胶电泳(图1A),载体和插入片段的条带大小均符合预期。将连接产物转化生长后,挑取多个单克隆提取质粒进行测序,通过与模板序列比对,成功构建目标序列pComb3H-BCMA-scFv,制备BCMA-scFv噬菌体。用酶切单位依次增加的牛凝血酶量0 U,2 U,4 U,6 U,8 U分别酶切100 μL表面展示BCMA-scFv的噬菌体,进行SDS-PAGE电泳和Western Blot(图1B、图1C),目标蛋白条带大小符合预期,随着酶切单位增加,目标蛋白浓度增加,酶切效果越好。

A:载体和插入片段的DNA凝胶电泳图;B:SDS-PAGE检测thrombin酶切噬菌体展示的BCMA-scFv; C:Western Blot检测thrombin酶切噬菌体展示的BCMA-scFv。图1 质粒的构建及噬菌体展示的BCMA-scFv检测图
2.2 包被BCMA抗原通过ELISA和测试洗脱噬菌体滴度检测牛凝血酶洗脱效果
用链霉亲和素孔板包被生物素化的BCMA抗原,按照ELISA的检测方法,在实验孔加入0 U,2 U,4 U,6 U,8 U不同牛凝血酶单位的洗脱液,测试洗脱液的噬菌体滴度(图2A),同时加入TMB显色液后显色于450 nm处读取吸光度(图2B)。如图2A、图2B所示,在包被BCMA抗原条件下,使用thrombin酶切洗脱的洗脱噬菌体滴度约是不使用thrombin酶切噬菌体滴度的10倍以上,具有显著性差异,按照节约试剂和效果最佳的原则,确定thrombin酶切单位2 U为最适用量。通过对噬菌体溶液进行梯度稀释来改变噬菌体浓度,对稀释后的噬菌体溶液进行ELISA检测,thrombin酶切洗脱后,检测洗脱液的噬菌体滴度和TMB显色后的OD450值(图2C、图2D)。实验组与对照组的噬菌体滴度相差10倍即为出现显著性差异,由图2C中可以看到,实验组的噬菌体滴度均与对照组具有显著差异,说明牛凝血酶可特异性洗脱表面展示BCMA-scFv的噬菌体,效果良好。
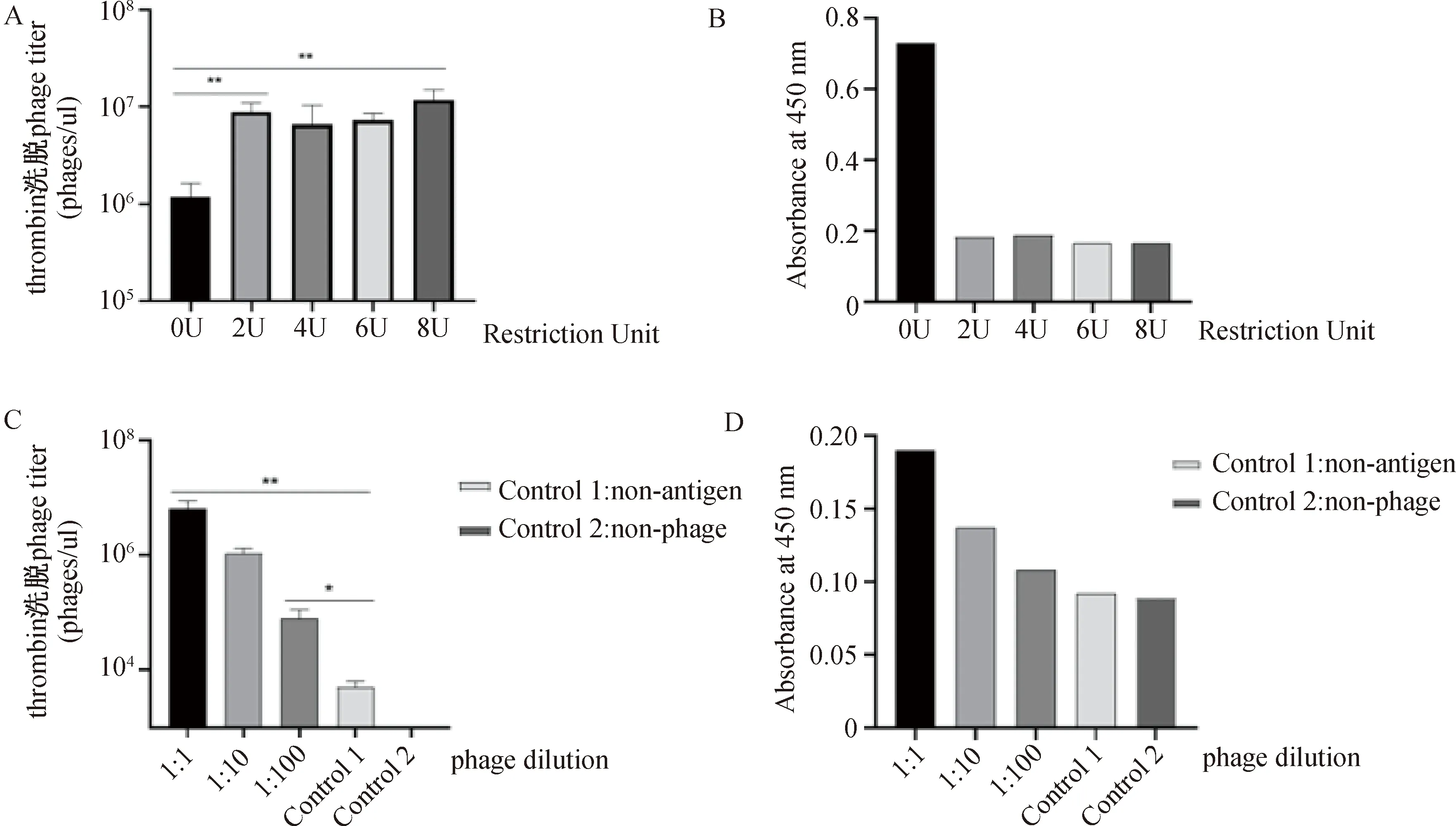
A:包被BCMA抗原通过ELISA确定该条件下最适酶切用量;B:包被BCMA抗原通过测试噬菌体滴度确定该条件下最适酶切用量;C:包被BCMA抗原通过ELISA检测thrombin洗脱效果;D:包被BCMA抗原通过测试噬菌体滴度检测thrombin洗脱效果。图2 包被BCMA抗原后,不同条件下检测thrombin酶切的噬菌体滴度和OD450
2.3 包被anti-Flag抗体通过ELISA和测试洗脱噬菌体滴度检测牛凝血酶洗脱效果
包被鼠源anti-Flag单抗,加入梯度稀释的噬菌体溶液,反应后在各实验孔中分别加入牛凝血酶单位依次增加0 U,2 U,4 U,6 U,8 U的洗脱液,按照ELISA的检测方法,先测定洗脱液的噬菌体滴度,然后加入TMB显色液显色后于450 nm处读取吸光度(图3A、图3D)。在包被anti-Flag抗体条件下,使用thrombin酶切洗脱的洗脱噬菌体滴度约是不使用thrombin酶切噬菌体滴度的100倍(10倍以上),具有显著性差异,按照节约试剂和效果最佳的原则,确定thrombin酶切单位2 U为最适用量。将噬菌体溶液梯度稀释,thrombin酶切洗脱后,实验组的噬菌体滴度均是对照组的1 000倍以上(出现10倍差距即为显著性差异),具有显著差异,说明牛凝血酶可特异性洗脱表面展示BCMA-scFv的噬菌体,效果良好。

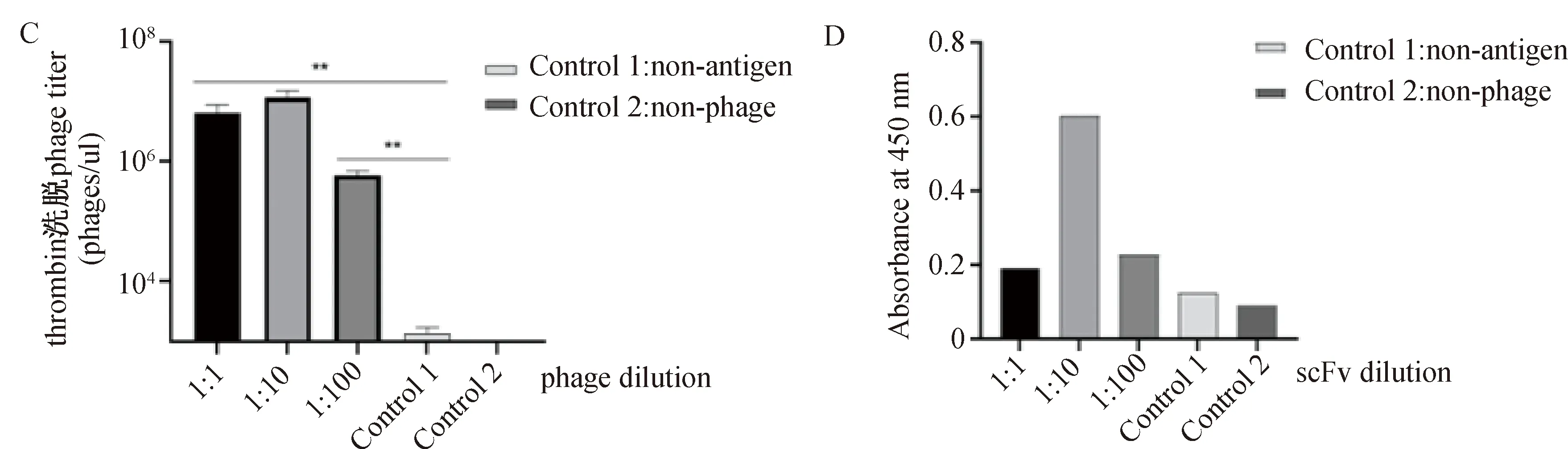
A:包被anti-Flag抗体通过ELISA确定该条件下最适酶切用量;B:包被anti-Flag抗体通过测试噬菌体滴度确定该条件下最适酶切用量;C:包被anti-Flag抗体通过ELISA检测thrombin洗脱效果;D:包被anti-Flag抗体通过测试噬菌体滴度检测thrombin洗脱效果。图3 包被anti-flag抗体后,不同条件下检测thrombin酶切的噬菌体滴度和OD450
2.4 通过模型筛选分析牛凝血酶特异性洗脱效果
将相同酶切位点的B7-H3-scFv基因与载体pComb3H连接到一起,载体和插入序列的条带大小如图4A所示,用得到的测序正确的pComb3H-B7-H3-scFv基因制备噬菌体。用链霉亲和素酶标板包被BCMA抗原,在B7-H3-scFv噬菌体溶液中混入梯度稀释的BCMA-scFv噬菌体,并按照ELISA的检测方法,每孔加入2 U单位的牛凝血酶洗脱特异性结合的噬菌体,收集的洗脱液感染XL1-Blue菌液,测定噬菌体的滴度,确定从混合噬菌体溶液中筛选出特异性结合BCMA的噬菌体的筛选轮数和效果。图4B所示为在一轮模型筛选后,与对照组相比,从展示B7-H3-scFv的噬菌体混合物中富集展示BCMA-scFv噬菌体高达2 000倍。分别从3组筛选组中挑选单克隆测序,经过序列的分析和对比后发现目标BCMA-scFv出现比例分别为100%,83.3%,16.7%与筛选富集度相符(图4C)。这些结果表明,在用于构建噬菌体抗体库的质粒上添加thrombin酶切位点,筛选过程采用特异性洗脱的方法,可以增加目标噬菌体的富集度,提高筛选目标特异性抗体的效率。
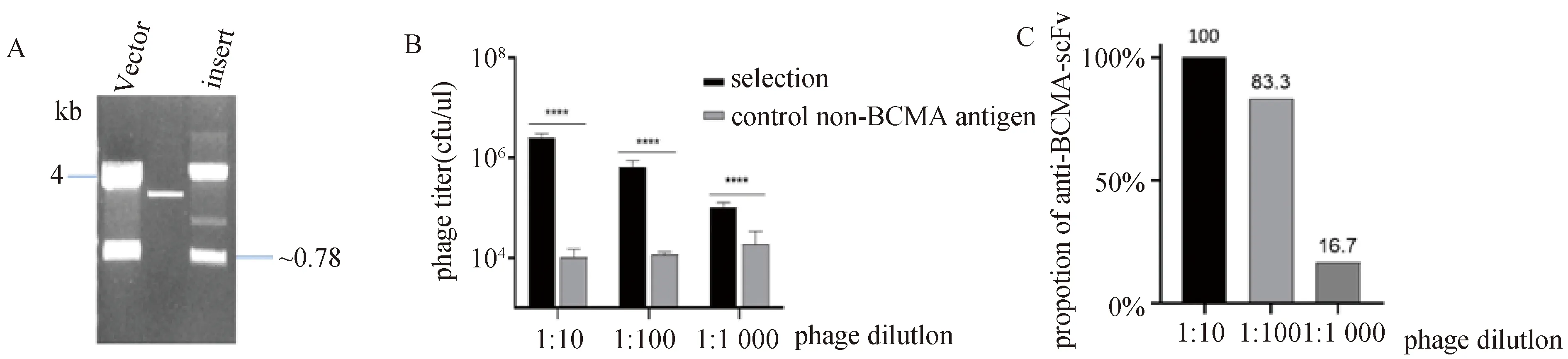
A:模型筛选pComb3H-B7-H3-scFv质粒构建图;B:不同混合比例下表面展示BCMA-scFv的目标噬菌体富集度;C:不同混合比例下目标BCMA-scFv比例。图4 模型筛选分析牛凝血酶特异性洗脱的效果图
3 讨论
噬菌体展示技术自1985年被创建至今已日趋完善,2018年由于该技术在抗体药物领域的贡献,该技术的发明人被授予诺贝尔化学奖,更推动了噬菌体技术的发展。噬菌体筛选技术不仅能筛选出高亲和力和特异性结合的单克隆抗体,也能筛选出不同形式的抗体,如scFv[13],Fab[14]等,应用价值极高。
各种不同的抗体基因所表达的多肽展示在噬菌体上,即可构建为噬菌体抗体库。若要在抗体库中筛选出特异性结合强的抗体,要先选择恰当的载体。本实验中使用噬菌粒载体,可避免融合蛋白的多价展示,实现更高的转化效率[15],主要包括3个关键要素(i)用于选择和繁殖质粒的抗生素标记(ii)编码scFv-G3P融合蛋白的基因(iii)滚环扩增和产生能够包装成噬菌体的(+)DNA链所需的M13染色体区域(噬菌体复制起点)。其次要选择合适的筛选方法,并根据每一轮筛选的结果进行优化。利用该技术,可以快速获得亲和力高的抗体。但是,筛选所用的相应抗原是细胞表面上抗原复合物或分子的一部分,包含不同的表位,噬菌体展示筛选通常仅产生针对抗原线性表位的抗体片段,为了富集和分离特异性抗体,通常需要多轮选择。最高亲和力结合的抗体片段或噬菌体在筛选过程中具有生长优势,因此,抗弱表位的抗体表位很少被选择,会导致筛选轮次中潜在结合物的多样性丧失,阻碍对亲和力较低的抗体的选择[16]。其次感染后,来自辅助噬菌体的野生型G3P蛋白与编码scFv-G3P融合蛋白的噬菌粒竞争并入噬菌体,90%的噬菌体群体不显示任何融合蛋白,并且带有融合蛋白的绝大多数噬菌体颗粒仅包含一个拷贝[17],这大大增加了筛选过程中非特异性结合的噬菌体数量,使得筛选轮次增加,筛选效率降低,难度加大。
因此,利用噬菌体展示技术如何快速高效并有效的筛选出目标抗体是我们一直努力的目标。在这里,我们提出了一种应用于噬菌体抗体库筛选的质粒构建新方法,目的是使噬菌体展示筛选效率提高,同时采用该质粒建立1种模型筛选方法。在融合噬菌粒scFv-PIII间引入牛凝血酶(thrombin)酶切位点,通过蛋白水解裂解,从而导致非特异性洗脱噬菌体的背景下降。该方法的创新在于:1、相对于强酸强碱非特异性洗脱方式,该方法洗脱液更温和、对大肠杆菌无害。虽然噬菌体在处于相当极端的环境中后仍然具有感染性,但采用酸碱中和非特异性洗脱的方式,对于噬菌体感染能力仍有不确定影响,该方法能够避免削弱噬菌体感染能力;2、降低每轮筛选过程中野生型噬菌体背景。牛凝血酶作用于scFv-PIII结合位点,不同于非特异性洗脱方式作用于抗原抗体结合位点,野生型噬菌体依然结合于链霉亲和素板,而洗脱液中含有筛选出的特异性噬菌体;3、构建的噬菌体抗体库模型筛选质粒可以作为实施单链抗体体外亲和力成熟模板,提高人源化单链抗体(scFv)亲和力。同时,我们采用该质粒建立1种模型筛选方法,目的是从表面展示抗B7-H3单链抗体的噬菌体混合物中高效筛选目标噬菌体,减少筛选轮次仅一轮即可高效富集,得到目标抗体,筛选方法优越。新的抗体库和筛选方法构建成功后,可以以此为基础高效筛选多种抗原的特异性抗体。
猜你喜欢
山东医药(2023年23期)2023-09-08 18:33:05
昆明医科大学学报(2022年2期)2022-03-29 00:51:20
中国动物传染病学报(2021年3期)2021-07-21 03:19:28
成都医学院学报(2021年2期)2021-07-19 08:35:20
中国乳品工业(2018年10期)2018-11-16 08:19:24
天然产物研究与开发(2018年10期)2018-11-06 07:43:50
上海故事(2015年13期)2016-01-22 13:25:09
广东海洋大学学报(2015年3期)2015-12-22 10:05:26
伴侣(2015年5期)2015-09-10 07:22:44
沈阳医学院学报(2014年4期)2014-12-27 13:44:32
