
Weighted single-step GWAS and RNA sequencing reveals key candidate genes associated with physiological indicators of heat stress in Holstein cattle
2022-12-20 09:41HanpengLuoLirongHuLuizBritoJinhuanDouAbdulSammadYaoChangLonggangMaGangGuoLinLiuLiweiZhaiQingXuandYachunWang
Hanpeng Luo,Lirong Hu,Luiz F.Brito,Jinhuan Dou,Abdul Sammad,Yao Chang,Longgang Ma,Gang Guo,Lin Liu,Liwei Zhai,Qing Xu and Yachun Wang*
Abstract Background:The study of molecular processes regulating heat stress response in dairy cattle is paramount for developing mitigation strategies to improve heat tolerance and animal welfare.Therefore,we aimed to identify quantitative trait loci (QTL) regions associated with three physiological indicators of heat stress response in Holstein cattle,including rectal temperature (RT),respiration rate score (RS),and drooling score (DS).We estimated genetic parameters for all three traits.Subsequently,a weighted single-step genome-wide association study (WssGWAS) was performed based on 3200 genotypes,151,486 phenotypic records,and 38,101 animals in the pedigree file.The candidate genes located within the identified QTL regions were further investigated through RNA sequencing (RNA-seq) analyses of blood samples for four cows collected in April (non-heat stress group) and four cows collected in July (heat stress group).Results: The heritability estimates for RT,RS,and DS were 0.06,0.04,and 0.03,respectively.Fourteen,19,and 20 genomic regions explained 2.94%,3.74%,and 4.01% of the total additive genetic variance of RT,RS,and DS,respectively.Most of these genomic regions are located in the Bos taurus autosome (BTA) BTA3,BTA6,BTA8,BTA12,BTA14,BTA21,and BTA24.No genomic regions overlapped between the three indicators of heat stress,indicating the polygenic nature of heat tolerance and the complementary mechanisms involved in heat stress response.For the RNA-seq analyses,2627 genes were significantly upregulated and 369 downregulated in the heat stress group in comparison to the control group.When integrating the WssGWAS,RNA-seq results,and existing literature,the key candidate genes associated with physiological indicators of heat stress in Holstein cattle are: PMAIP1,SBK1,TMEM33,GATB,CHORDC1,RTN4IP1, and BTBD7.Conclusions:Physiological indicators of heat stress are heritable and can be improved through direct selection.Fiftythree QTL regions associated with heat stress indicators confirm the polygenic nature and complex genetic determinism of heat tolerance in dairy cattle.The identified candidate genes will contribute for optimizing genomic evaluation models by assigning higher weights to genetic markers located in these regions as well as to the design of SNP panels containing polymorphisms located within these candidate genes.
Keywords: Climatic resilience,Dairy cattle,QTL mapping,RNA sequencing,WssGWAS
Graphical Abstract
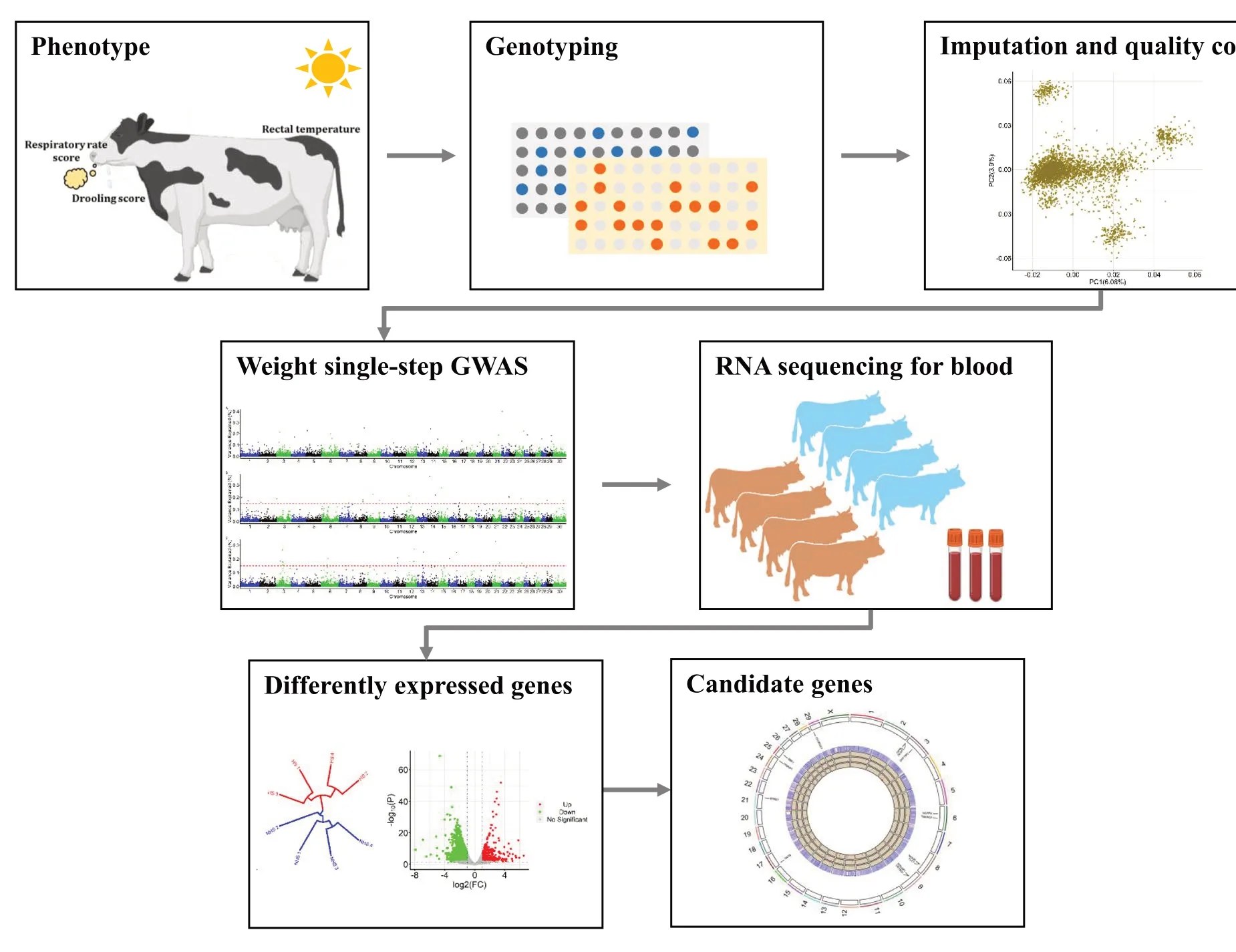
Introduction
A global warming trend has been observed over the last century [1],which has unfavorable effects on livestock production.Thus,heat stress is becoming a major challenge facing the global dairy industry [2].Heat stress is the result of the sum of external forces acting on an animal that evokes a series of behavioral and physiological responses,including sweating,higher respiration rate,vasodilation with increased blood flow to skin surface,reduced metabolic rate,decreased dry matter intake,and altered water metabolism [3].During periods of heat stress the hypothalamic-pituitary-adrenal (HPA)and the sympathetic-adrenal-medullary (SAM) axis are activated for maintaining homeostasis in response to stressful stimuli [4].Under chronic stress,cortisol secretion associated with immune suppression [5,6] leading to the animal becoming more susceptible to disease and immune challenges [7].Heat stress is shown to severely alter the welfare,as well as productive and reproductive performance of dairy cows [8].Due to the thermal hysteresis existing [9] for milk yield in dairy cattle,physiological performance traits are better indicators for monitoring heat load of individuals.We have established two scoring systems for respiration rate and salivation (drooling)in addition to measuring rectal temperature in Holstein population [10].Respiration rate score (RR),rectal temperature (RT),and drooling score (DS) are lowly heritable traits controlled by numerous quantitative trait loci(QTL),each with a small effect,thereby making it more difficult to obtain fast genetic progress for these traits.
Genome-wide association studies (GWAS) are used to discover genomic regions associated with phenotypes of interest [11],and consequently,biological processes in which they are involved in.For instance,Luo et al.[12] identified candidate genes related to RT by using single-SNP regression GWAS.Subsequently,gene expression analyses were performed to validate the key functional genes identified[12].A recent method known as single-step GWAS[13] is becoming the gold standard method for GWAS as it enables the simultaneous integration of genomic,phenotypic,and pedigree information in a single analysis.ssGWAS has been widely used for scanning QTLs associated with complex traits in dairy cattle such as milk yield and composition traits in Holstein cattle[14,15];fertility and reproductive disorders [16];and heat tolerance [17].Furthermore,the combination of GWAS and transcriptomics has been shown to significantly enhance the understanding of the genetic architecture of complex traits [18].Understanding how genetic variation shapes the phenotypic variability of complex traits requires the identification of such genetic polymorphisms through GWAS combined with functional evaluation of the key candidate genes through RNA sequencing (RNA-seq).RNA-seq is a technique that evaluates the quantity and sequences of RNA in a sample based on next generation sequencing (NGS) tools.RNA-seq analyzes the full transcriptome,indicating which of the genes encoded in the DNA are turned on or off and to what extent [19].To our best knowledge,no study has combined weighted single-step GWAS (WssGWAS) and RNA-seq for the determination of candidate genes and genetic variants associated with physiological indicators of heat stress in dairy cattle.Thus,the main objectives of this study were to: 1) identify genomic regions associated with RT,RR,and DS in Holstein cattle based on WssGWAS;2) search for candidate genes in the QTL regions that explain greater proportions of the total additive genetic variance of each trait;and,3) utilize RNA-seq and gene network analyses to investigate the biological processes shared by the candidate genes identified for the three indicators of heat tolerance.
Material and methods
Phenotype,pedigree and genotypic data
A total of 69,837 (RT),40,760 (RR),and 40,889 (DS)phenotypic records were collected in 15,303 lactating Holstein cows from 2013 to 2020.The process of data collection,scoring protocol of RR and DS,and farm management have been described in details by Luo et al.[10].The descriptive statistics for the phenotypic data and environmental index (temperature-humidity index,THI) are presented in Table 1.Each lactating cow was recorded twice a day for two consecutive days(07:00–11:00 and 14:00–18:00).A small proportion of cows were measured in more than one lactation (i.e.,>4 records/cow).
The pedigree file contained 38,101 animals,spanning over three generations.A total of 3200 animals(3119 cows and 81 bulls) were genotyped using the Illumina 150 K Bovine Bead chip (Illumina,Inc.,San Diego,CA,USA),which contains 139,377 single nucleotide polymorphism (SNPs).The individuals with call rate higher than 0.9 were kept and the call rate of genotyped individuals is 0.977 before imputation.All of the genotyped animals were included in a genotype imputation analyses for imputing the missing SNPs.This was done using the Beagle5.1 software [20].Genotype quality control kept SNPs with: minor allele frequency (MAF) greater than 0.05,no extreme departure from Hardy-Weinberg equilibrium (P-value greater than 10−6),known chromosome and genome position,and located in the autosomal chromosomes.After the quality control,114,766 SNPs and 3200 animals were kept for further analyses.
Statistical analyses
A multiple-trait model was employed to estimate variance and covariance components,which can be described as follows:
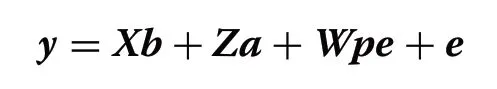
whereyis the vector of phenotypic records (RT,RR,and DS);bis the vector of systematic effects includingfarm-year (for RT) or farm-year-scoring person (for TT and DS),parity (1,2,or 3+),lactation stage (days in milk 1–50,51–100,101–150,151–200,201–250,251–300,or >300),milking status (data collected before milking,after milking,or unknown),and THI (as a continuous covariable in the model fitting a linear regression);ais the vector of random additive genetic effects;pis the vector of random permanent environmental effects;eis the vector of random residuals;andX,Z,andWare the incidence matrices linking phenotypic records tob,a,andpe,respectively.The model assumptions are:
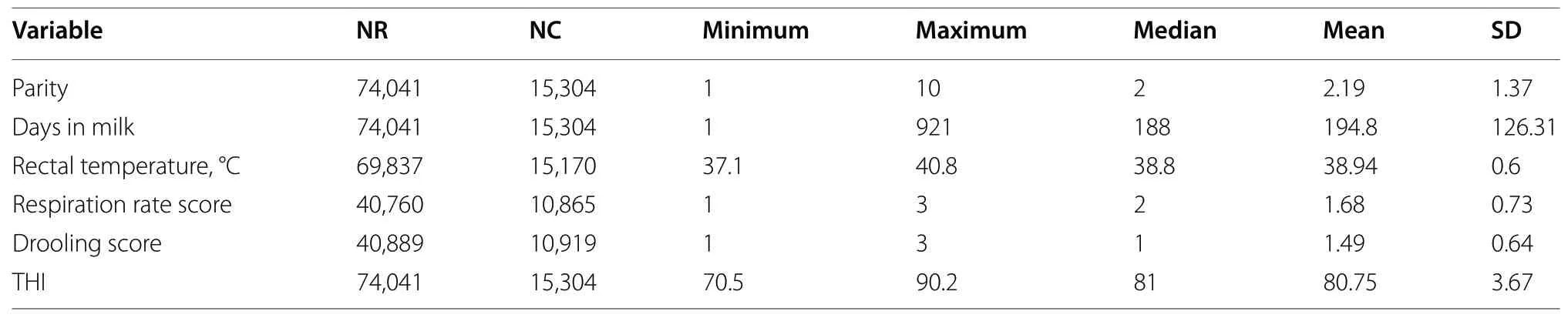
Table 1 Descriptive statistics of phenotypic records and environmental variables used in the analyses
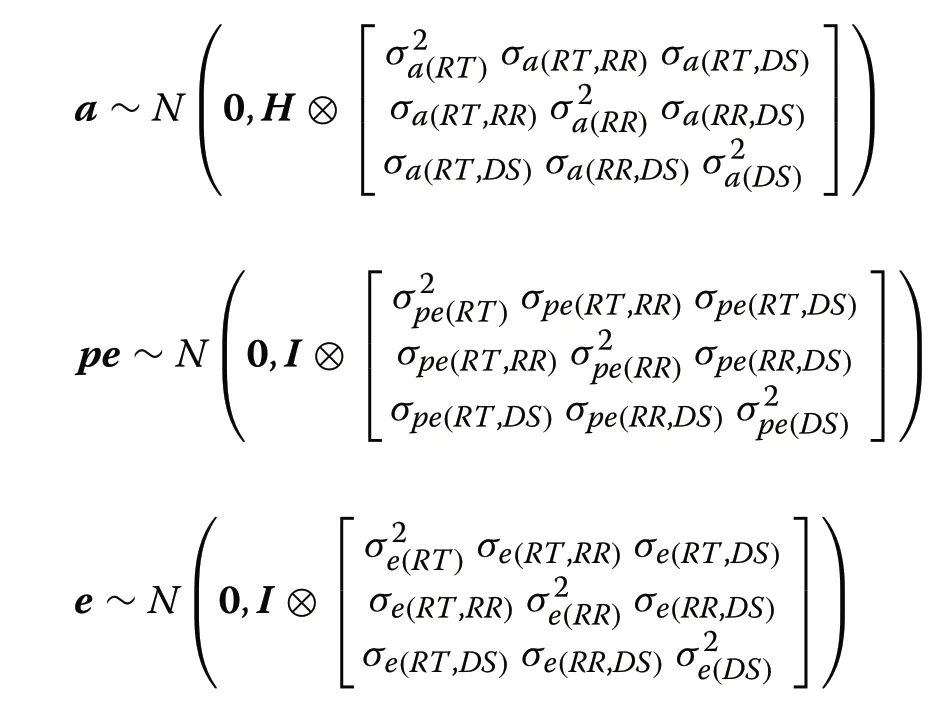
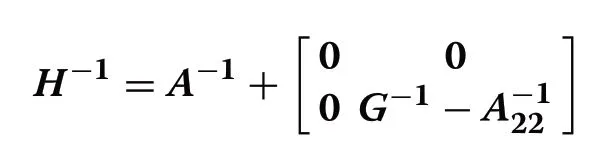
whereAis the numerator relationship matrix based on pedigree for all animals;A22is the numerator relationship matrix for genotyped animals.TheGmatrix was computed as [22]:
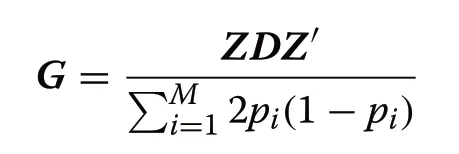
whereZis a matrix of gene content adjusted for allele frequencies (0,1,or 2 for aa,Aa,and AA,respectively);Dis a diagonal matrix of weights for SNP variances (initiallyD=I);Mis the number of SNPs,andpiis the minor allele frequency of theithSNP.Variance components were estimated using the average information-restricted maximum likelihood (AI-REML) procedure implemented in the AIREMLF90 package from the BLUPF90 family programs [23].
Weighted single-step genome-wide association study(WssGWAS)
The estimates of SNP effects and weights for the Wss-GWAS analyses (three iterations) were obtained according to Wang et al.[24].The weight for each SNP was calculated as:[22],whereiis theithSNP.The percentage of the total additive genetic variance explained by theithregion was calculated as:

Whereaiis genetic value of theithregion that consists of contiguous 10 SNPs,is the total additive genetic variance,Zjis a vector of gene content of thejthSNP for all individuals,and ˆujis the marker effect of thejthSNP within theithregion.
Candidate genes detection and functional enrichment analyses
Genomic windows of 10 consecutive SNPs that explained 0.15% or more of the total additive genetic variance,based on the WssGWAS analyses,were considered to be associated with the studied traits.A Manhattan plot was created using the R software [25].Genes were annotated on the basis of the starting and ending coordinates of each window (using the ARS-UCD1.2 assembly as the reference genome;GCA_002263795.2) by using the R package ‘Biomart’ of Ensembl [26].Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways and Gene Ontology (GO) terms were enriched via the “clusterProfiler” package [27].
RNA sequencing
Eight primiparous cows with DIM ranging from 135 to 144 d (mid-lactation and pregnant) were selected from 3200 genotyped individuals for RNA-seq.Blood samples were collected via the coccygeal venipuncture in the Spring (4 cows in April during the thermoneutral period with average daily temperature lower than 25 °C) and Summer (4 cows in July during heat stress period with average daily temperature higher than 25 °C) seasons,following the RT,TT,and DS recording on the same day.RT,TT,and DS were collected after blood samples collected to reduce artificial stimulation.The experimental design in current study is consisted with other previous research investigating heat stress by RNA-seq [28,29].The RNA was isolated from blood according to the manufacturer’s instructions of the TRIzol Reagent method[30].The RNA concentration and quality were assessed using the Equalbit RNA BR Assay Kit (Invitrogen,CA,USA) and Nanodrop 2000 (Thermo,Massachusetts,USA).RNA integrity was detected by 1% agarose gel electrophoresis and used for library construction with 28S/18S >1.For RNA-seq library,2 μg of RNA was used for purification and fragment using NEBNext Poly(A)mRNA Magnetic Isolation Module (Cat No.E7490S,New England Biolabs Ltd.,Hitchin,Herts,UK) then followed by cDNA library with NEBNext Ultra RNA Library Prep Kit for Illumina (Cat No.E7530S,New England Biolabs Ltd.,Hitchin,Herts,UK).All libraries were quantified by Equalbit DNA BR Assay Kit (Invitrogen,CA,USA) and pooled equimolarly.They were finally submitted for sequencing using the NovaSeq 6000 System (Illumina,Inc.,San Diego,CA,USA) which generated 150 bases paired-end reads.
Differential expression and functional analyses
The quality of the sequencing reads was evaluated using the FastQC software (v0.11.9) and global trimming using the Fastp [31].All clean reads were mapped to the bovine genome of version ARS-UCD1.2 using the software STAR [32],and a Picard query [33] was carried out to eliminate duplicates.We investigated population structure through a principal component analysis (PCA) implemented in the PLINK software[34].For differential expression gene screening,exact test based on quantile-adjusted conditional maximum likelihood (qCML) was performed using the edgeR [35]R package with criteria fold change ≥ 2 and 0.05 for the alpha of false discovery rate (4 samples vs.4 samples).The heatmap was constructed using the pheatmap R package [36].
Results
Genetic parameter estimates
Estimates of variance components for RT,RR,and DS are shown in Table 2.The heritability estimates ranged from 0.03 (DS) to 0.06 (RT),with repeatability estimates ranging from 0.12 (DS) to 0.19 (RT),indicating the large environmental influence in these indicators of heat stress.Statistically significant genetic correlations were observed for RT with RR (0.22) and RR with DS (0.21).
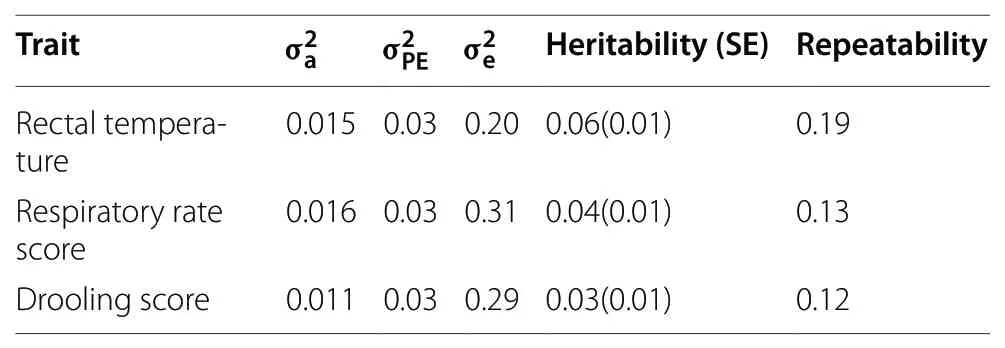
Table 2 Genetic parameters,heritability,and repeatability for the evaluated physiological traits under heat stress
QTL mapping and trait-related gene identification
Figure 1 presents the genetic variance of each genomic window after performing WssGWAS with three iterations.Fourteen,19,and 20 genomic regions reached the predefined threshold (0.15%) and they explained 2.94%,3.74%,and 4.01% of the total additive genetic variance for RT,RR,and DS,respectively.Most of these genomic regions are located in theBos taurusautosome (BTA) BTA3,BTA6,BTA8,BTA12,BTA14,BTA21,and BTA24.However,we found no overlapping genomic regions between the three physiological traits in this study.The detailed information about these genomic regions related to the three physiological traits and candidate genes are presented in Table 3.Many windows with small effects regulated RT,RR,and DS co-occurred,indicating that these three physiological indicators of heat stress are highly polygenic traits.A total of 54 protein-coding genes (27,14,and 13 protein-coding genes located by the genomic region associated with RT,RS,and DS,respectively) were annotated in these genomic regions according to the Ensembl database.The GO enrichment and KEGG pathway analyses of the 54 protein-coding genes for the three physiological traits revealed 68 significant GO terms (Additional file 1:Fig.S1) including biological process as well as molecular functions and two significant KEGG pathway (purine metabolism and thiamine metabolism).
General features and genetic background of cows sampled for RNA sequencing analysis
The environmental THI and physiological performance(RT,RR,and DS) parameters during the two sample collection periods (April is the thermoneutral season in Beijing,named non-heat stress group,while July is the heat stress season,named heat stress group) were significantly different.The milk yield of the cows in the nonheat stress group (41.48 ± 3.52) was significantly higher than the cows in the heat stress group (35.25 ± 3.71).The description of environmental THI,physiological performance,and milk yield during periods of blood samples collecting are shown in Table 4.The genetic relationship of the dairy population evaluated is presented in Fig.2,which shows a diverse genetic background of the cows(red and blue dots) sampled for the RNA-seq analysis.
Identification of differentially expressed mRNAs by sequencing
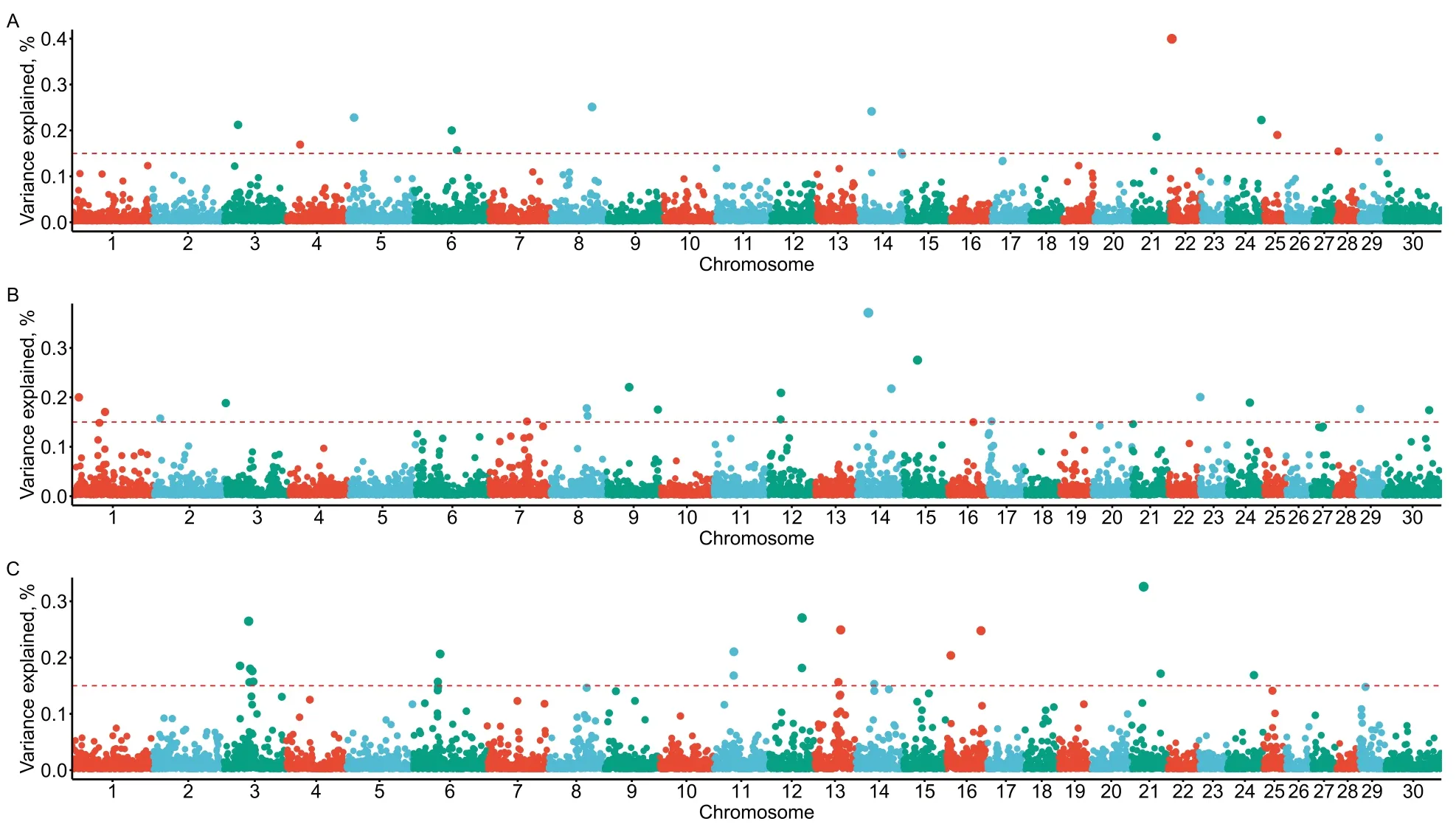
Fig.1 Proportion of the total additive genetic variance of 10-SNP genomic windows based on the weighted single-step genome association studies.A The Manhattan plot for rectal temperature.B The Manhattan plot for respiration rate score.C The Manhattan plot for drooling score.The red dashed lines represent the threshold 0.15% of the total additive genetic variance
There is a significant difference between the heat stress(HS) group and non-heat stress (NHS) group for RNAseq counts based on principle component analysis and clustering structure (Fig.3).The analysis of the differently expressed genes (DEGs) were detected based on the quantile-adjusted conditional maximum likelihood method.A total of 2627 significantly downregulated genes were found and 369 upregulated genes in the NHS group compared to HS group (Additional file 2: Table S1).The GO enrichment and KEGG pathway analysis of DEGs revealed 21 significant GO terms including Biological Process,Cellular Component,and Molecular Function and 86 significant KEGG pathways (Additional file 3:Fig.S2).There were 14 genes identified based on the WssGWAS that were also DEGs.Table 5 shows that two of the significant common genes are upregulated and the other ones are downregulated.Figure 4 shows the heatmap of the mRNA expression of these 14 genes using hierarchical cluster of log2 from the relative normalized expression in bovine blood transcriptome.
Discussion
The estimation of genetic parameters and detection of genomic regions for three physiological indicators of heat stress in Holstein cattle contributes to further understanding of the genetic architecture of heat tolerance in cattle.The heritability estimates for the three traits are low (0.03–0.06) in the population evaluated but statistically different than zero.The variance component results utilizing theHmatrix are similar to earlier estimates for the same traits using the numerator relationship matrix based on pedigree (Amatrix;[10],indicating that the size of the datasets used is large enough to accurately estimate genetic parameters for these three traits.The heritability estimated for RT in 1695 lactating Holstein cows during heat stress (0.17 ± 0.13;[37]) and in 3396 straight-bred and crossbred Romosinuano,Brahman,and Angus cattle(0.19 ± 0.03;[38] using bivariate models with coat score are higher than the estimates observed in our population.However,their accuracies of estimation are lower due to their smaller datasets.
For low-heritability traits,substantial genetic progress can be achieved by increasing intensity and accuracy of selection [39,40].Subsequently,genomic selection could significantly speed up the rates of genetic progress[41] by improving accuracy of selection and reducing generation interval.In our earlier study [12],single-SNP regression GWAS was performed for RT in a subset of the current population including 7598 Chinese Holstein cattle with 1114 genotyped cows.Ten SNPs(located on BTA3,BTA4,BTA8,BTA13,BTA14,and BTA29) were found to be significantly associated with RT and five positional candidate genes were identified(SPAG17,FAM107B,TSNARE1,RALYL,andPHRF1).In the present study we identified 53 trait-related genomic regions and 53 protein-coding genes through WssGWAS.Contrary to the single-SNP GWAS,moregenomic regions were captured via WssGWAS (e.g.,[42,43],showing that WssGWAS can be more successful in detecting QTL.According to study of simulation [42],single-step GWAS (based on single-step genomic best linear unbiased prediction) could take care of correction for population structure in GWAS like other mixed linear models (e.g.EMMAX).Comparing to the method of WssGWAS,BayesB appears to overly shrink regions to zero,while overestimating the amount of genetic variation attributed to the remaining SNP effects in chicken population [24].We compared two scenarios (weighting step including re-estimated GEBV and weighting step only with re-calculated SNP effect [24]) along with the inclusion of times of iteration for weighting (up to 5 iterations,results not shown).Although,the process of WssGWAS with only re-calculated SNP effects or more than three iterations resulted in too much noise (results not shown),which is consistent with prior studies based on simulated data in dairy cattle [44] and real data in broiler chickens [24].Additionally,it is critical to select the number of SNPs included in the sliding windows when performing (W)ssGWAS as wider windows could capture more genomic regions [45,46].The SNP chip used has an average inter-marker spacing of 26.07 kb,and the average distance between adjacent SNPs with linkage disequilibrium (r2) higher than 0.4 was 200 kb in the studied population.Therefore,we selected 10-SNP windows in linkage disequilibrium when scanning QTL related to the three physiological traits.
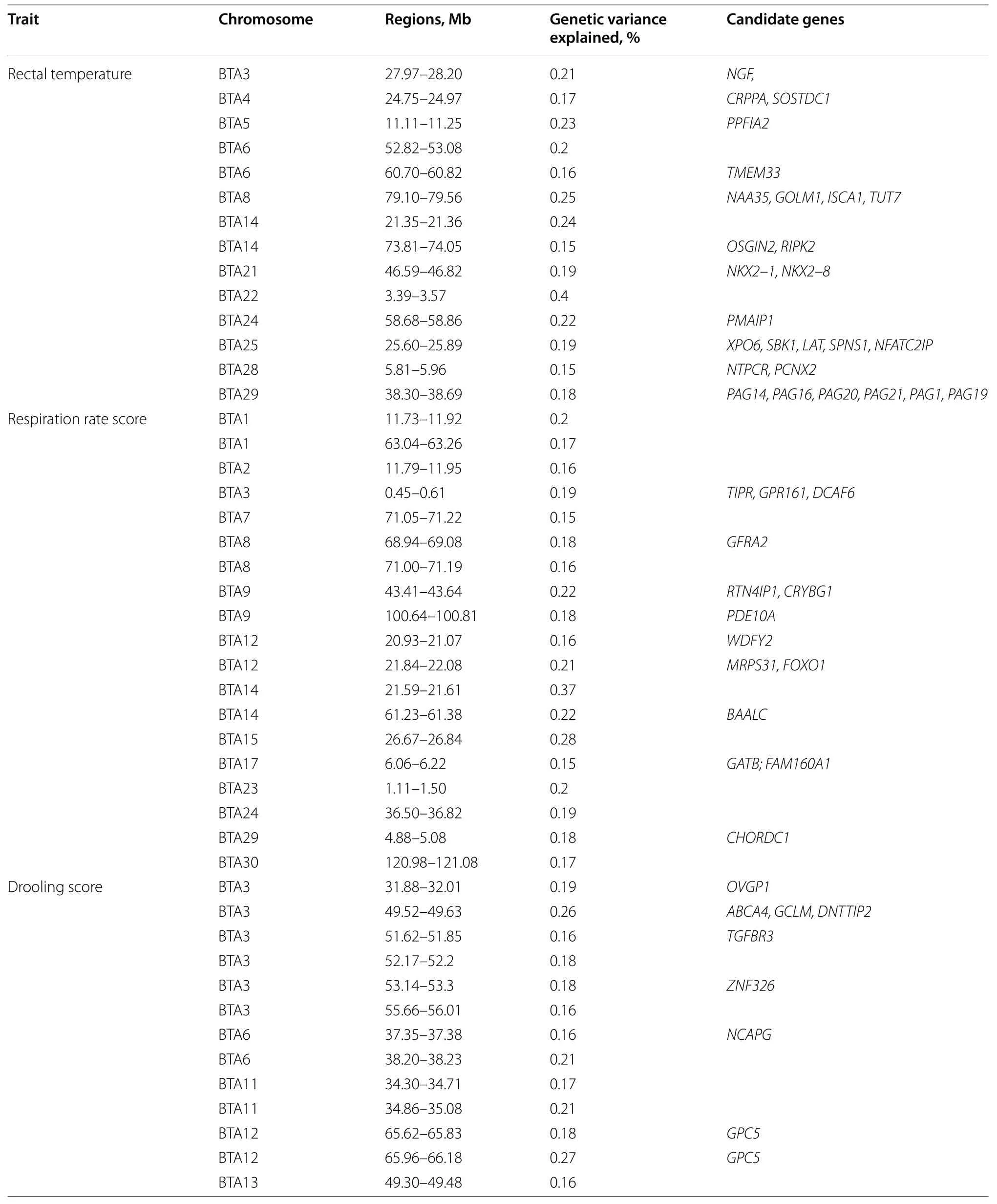
Table 3 Information of 10-SNP windows explaining more than 0.15% of the total additive genetic variance for rectal temperature,respiration rate score,and drooling score in Holstein cattle
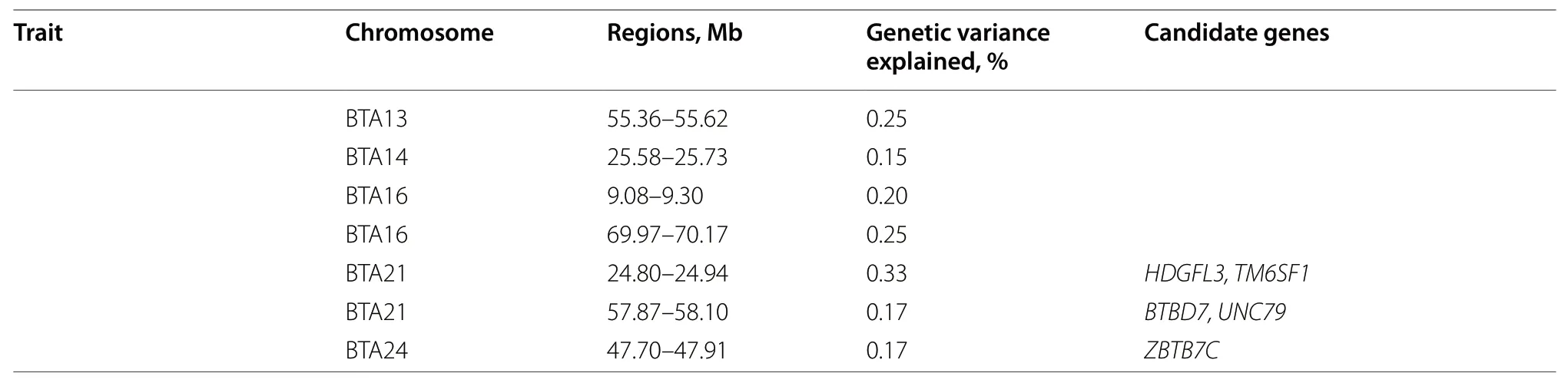
Table 3 (continued)
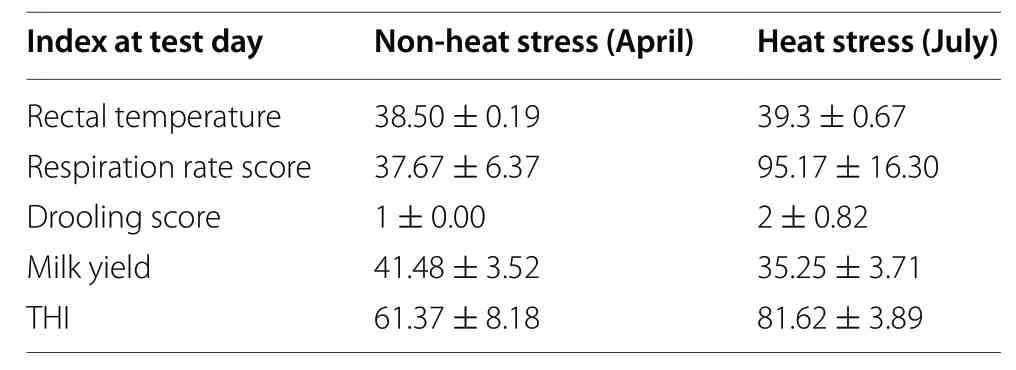
Table 4 Summary statistics for environmental THI,physiological performance,and milk yield in April (non-heat stress) and July(heat stress)
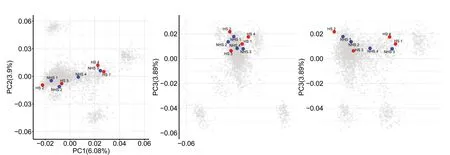
Fig.2
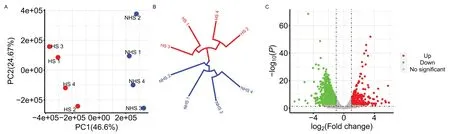
Fig.3 Cluster of heat stress and non-heat stress on the basis of read counts and volcano plot displaying differentially expressed genes.A,B Principle component analysis and dendrogram for samples collected under heat stress and non-heat stress on the basis of read counts.Red points in dot plot and red lines in dendrogram represent heat stress samples.Blue points in dot plot and blue lines in dendrogram represent heat stress samples.C Volcano plot showing significantly expressed genes.The red and green dots denote significantly downregulated and upregulated genes,respectively
RNA-seq analysis contributes to annotating new genes and splice variants,and provides cell-and context-specific quantification of gene expression.Research over the last few years has identified some of the physiological,metabolic,cellular,and molecular responses to heat stress in cattle[47,48].However,the level of gene expression depends on the physiological state of the organism and tissue analyzed.Cellular and transcriptomic adaptation of bovine granulosa cells were characterized to different heat stress intensities(39 °C,40 °C,and 41 °C) in-vitro [49].Several heat-responsive genes from different functional classes were identified and their associated pathways related to heat stress chaperons,cell death,apoptosis,hormonal synthesis,and oxidative stress.The expression of miRNAs in dairy cattle mammary gland under heat stress was investigated [50](483 known bovine miRNAs and 139 novel miRNAs were identified),which detected the heat-dependent differential modulation of miRNAs.Decreased expression of heat response-associated genes in blood during HS was also observed very recently in lactating cows [28].However,it was not considered that if metabolism of the milking stage from Spring to Summer directly affects the gene expression levels.In the current study of RNA-seq,we selected primiparous cows with similar DIM to avoid the effect of different lactation stage and the identified DEGs involved in the pathway of apoptosis,cellular senescence and autophagy,known to be affected by heat stress.
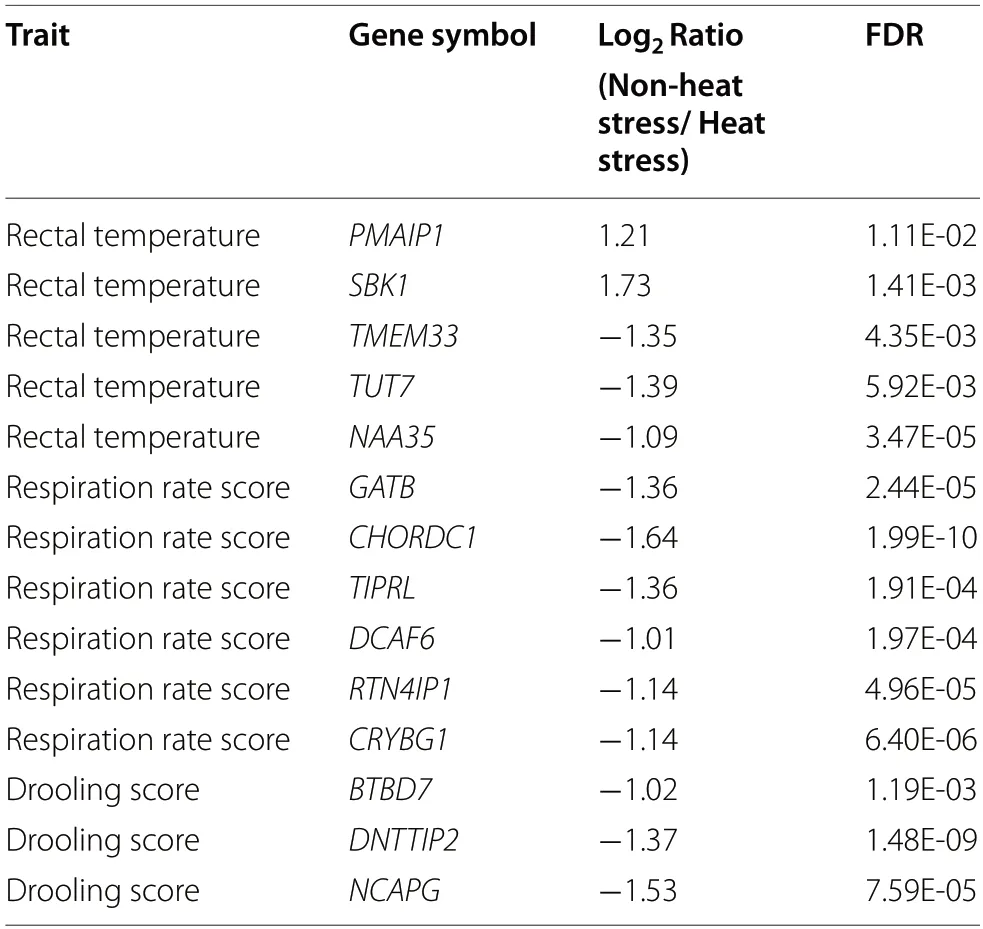
Table 5 List of overlapping genes between weighted singlestep genome-wide association analysis and RNA sequencing
Validation of the results of GWAS is an essential component of the experimental program.It could increase the level of trust held by animal breeders in genetic improvement,and is important as confirmation of a scientific hypotheses [51].There are many methods used to verify identified candidate genes/QTLs from GWAS results,e.g.,SNP chip analysis [52],qRT-PCR [12],or association analysis in another population [53].RNA-seq with the capabilities of high-throughput sequencing,could help us better understand the translation of genetic loci into biological mechanisms that underlie phenotypic expression of important traits [54].Fourteen genes identified in WssGWAS were significantly and differently expressed between cows in heat stress period and thermoneutral conditions.PMAIP1is a proapoptotic member of the BCL-2 protein family that acts as a proapoptotic sensitizer/de-repressor and regulates diverse cellular functions in autophagic cell death and metabolism [55,56].When comparing to heat shock of 38 °C and 43 °C,the expression ofPMAIP1was significantly and differentially upregulated in control (32 °C) mouse spermatogenic cells [57].Another upregulated gene during heat stress based on the differential gene analysis isSBK1,which has been reported as part of widespread expression pattern involved in the protection of cells from apoptosis induced by the viral infection in ovarian cancer cells and promoting the cellular survival [58].
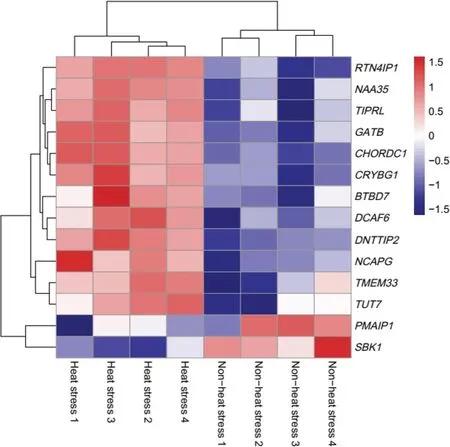
Fig.4 Heat map showing the relative normalized expression of the mRNA of 14 genes in eight individuals between period of April-non-heat stress and July-heat stress using hierarchical cluster.Genes with increased expression are shown in red while those with decreased expression are shown in blue
Five genes (TMEM33,GATB,CHORDC1,RTN4IP1,andBTBD7),out of the 12 significantly downregulated genes,were identified in prior studies involving heat stress/heat shock and cellular adaptive functions in the presence of stressors or disrupting molecular mechanisms.For instance,the overexpression ofTMEM33cause expression of endoplasmic reticulum stressinduced cell death signals increase,which leads to activation of the unfolded protein response signaling cascade and induction of an apoptotic cell death [59].The geneGATBwas identified to show significant transcriptional differences (downregulated gene) in response to heat shock where a temperature shift from 37 °C to 42 °C was accessed through the use of a microarray [60].TheGATBgene downregulation is shown to be responsible for the respiratory chain enzyme deficiencies [61],and thus disturbing the mitochondrial function and possesses complications for the much-needed energy intensive physiological mechanisms to dissipate incremental heat load in cattle.Furthermore,during the temperature increase phase,GATBcomposed an operon and was downregulated inClostridium botulinum(temperature shift from 37 °C to 45 °C over a period of 15 min;[62].GO annotations related toCHORDC1include Hsp90 protein binding,and this gene was associated with the regulation of heat stress and immune response processes in rats[63].Additionally,upregulation ofCHORDC1is shown to be primarily associated with dystrophic conditions[64].Therefore,it can be assumed that the downregulation ofCHORDC1is a positive adaptive sign of mechanisms involving thermotolerance.RTN4IP1(Reticulon 4 Interacting Protein 1) is included in the gene ontology of oxidoreductase activity and transferase activity.The proteins ofRTN4IP1were down-regulated at 26 °C compared to 18 °C in wild-type zebrafish [65].The expression ofRTN4IP1tend to decrease in the presence of stressors and decreased HSPs response,which again points towards a sort of possible homeostatic cellular response mechanisms initiated by the heat stressed dairy cow.BTBD7[BTB (POZ) domain-containing 7] is involved in a variety of biological functions [66].BTBD7has been shown to be induced by a matrix protein at sites of cleft progression and induce a transcription factor and suppress cell adhesion [67].Other studies describedBTBD7as a cell growth suppressor protein (ZNF238is expressed in postmitotic brain cells and inhibits brain tumor growth) and a promotor of angiogenesis [68].This function points out to the possible conservation mechanisms of heat stressed cows showing high energetic mechanisms directed at attaining thermo-neutrality and mechanisms of vasodilation of peripheral blood distribution network[69].Query of literature strongly support the involvement of the possible candidate genes proposed in this study,in the regulatory mechanisms directed at differential homoerotic and homeostatic mechanisms manifesting in various physiological modifications towards heat stress,as described in previous studies [70].Given the relevant support from prior research studies about the possible involvement in heat stress/heat shock response,the identification of genomic regions and candidate genes through WssGWAS,and biological validation through RNA-seq analyses indicate seven genes (PMAIP1,SBK1,TMEM33,GATB,CHORDC1,RTN4IP1,BTBD7) as likely playing an important role in heat stress response.Further studies should investigate these candidate genes in more depth and in more controlled on-farm or in-vitro experiments in response to more divergent environmental heat loads.
Conclusions
Physiological indicators of heat tolerance are heritable and can be improved through direct selection.We identified 54 candidate genes associated with rectal temperature,respiration rate score,and drooling score in Chinese Holstein cattle using WssGWAS.A validation experiment based on RNA-seq of blood samples in Holstein cattle provided evidence of their possible role in the physiological responses to heat stress.The identified candidate genes (PMAIP1,SBK1,TMEM33,GATB,CHORDC1,RTN4IP1,BTBD7) may provide knowledge for developing genomic evaluation models by assigning higher weights to genetic markers located in these regions or the development of SNP panels contained polymorphisms in these genes.
Abbreviations
RT: Rectal temperature;RR: Respiration rate score;DS: Drooling score;QTL:Quantitative trait loci;GWAS: Genome-wide association studies;RNA-seq: RNA sequencing;NGS: Next generation sequencing;WssGWAS: Weighted singlestep GWAS;THI: Temperature-humidity index;DIM: Days in milk;AI-REML: Average information-restricted maximum likelihood;KEGG: Kyoto Encyclopedia of Genes and Genomes;GO: Gene Ontology;DEGs: Differently expressed genes;HS: Heat stress;NHS: Non-heat stress season.
Supplementary Information
The online version contains supplementary material available at https:// doi.org/ 10.1186/ s40104-022-00748-6.
Additional file 1: Fig.S1.The 68 significant GO terms revealed by 54 protein-coding genes associated with the three physiological traits.
Additional file 2: Table S1.Differential gene expression between nonheat stress group and heat stress group.
Additional file 3: Fig.S2.The significant GO terms and pathways were enriched by Differential gene expression between non-heat stress group and heat stress group.
Acknowledgements
The authors acknowledge the Beijing Dairy Cattle Center and Beijing Sunlon Livestock Development Company Limited for providing access to the datasets used and enabling data collection at their farms.The authors are also grateful to the members of the COWINFO group at the China Agriculture University for helping to measure rectal temperature in a large number of animals,especially Lu Cao,Zezhao Wang,Xinyi Yan,Hetian Huang,Rui Shi,and Ye Wang.
Authors’ contributions
HL,LB,QX,and YW conceived the study.HL,LH,JD,YC and LM analyzed the data.HL wrote the first draft of the manuscript.LB,YW,AS,and LZ edited the manuscript.GG and LL participated in the data collection.All the authors read and approved the final version of the manuscript.
Funding
This research was funded by National Key R& D Program of China (2021YFD1200903,the earmarked fund for CARS36,the Program for Changjiang Scholar and Innovation Research Team in University (IRT_15R62).
Availability of data and materials
The data sets used during the current study are available from the corresponding author on reasonable request.
Declarations
Ethics approval and consent to participate
The study was approved by the ethical committee of the College of Animal Science and Technology,China Agricultural University,Beijing,China.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Author details
1Laboratory of Animal Genetics,Breeding and Reproduction,Ministry of Agriculture of China,National Engineering Laboratory of Animal Breeding,College of Animal Science and Technology,China Agricultural University,No.2,Yuanmingyuanxi Road,Haidian District,Beijing 100193,China.2Department of Animal Sciences,Purdue University,West Lafayette,IN 47907,USA.3College of Animal Science and Technology,Beijing University of Agriculture,Beijing 100096,China.4College of Animal Science,Xinjiang Agricultural University,Urumqi 830052,China.5Beijing Sunlon Livestock Development Company Limited,Beijing 100029,China.6Beijing Dairy Cattle Center,Beijing 100192,China.7College of Life Sciences and Bioengineering,Beijing Jiaotong University,Beijing 100044,China.
Received:14 December 2021Accepted:24 June 2022
 Journal of Animal Science and Biotechnology2022年6期
Journal of Animal Science and Biotechnology2022年6期
- Journal of Animal Science and Biotechnology的其它文章
- Toxicological effects of nanoselenium in animals
- The health benefits of selenium in food animals:a review
- The role of extracellular vesicles in animal reproduction and diseases
- The m6A methylation regulates gonadal sex differentiation in chicken embryo
- Integrative analysis of miRNA and mRNA profiles reveals that gga-miR-106-5p inhibits adipogenesis by targeting the KLF15 gene in chickens
- Genome-wide identification of functional enhancers and their potential roles in pig breeding