
Relapsing polychondritis with isolated tracheobronchial involvement complicated with Sjogren's syndrome: A case report
2022-12-19 08:06JunYanChenXiaoYanLiChenZong
World Journal of Clinical Cases 2022年19期
Jun-Yan Chen, Xiao-Yan Li, Chen Zong
Abstract
Key Words: Relapsing polychondritis; Tracheobronchial involvement; 18F-fluorodeoxyglucose positron emission tomography/computed tomography; Fiberoptic bronchoscopy; Case report
lNTRODUCTlON
Relapsing polychondritis (RP) is a systemic disease that primarily affects cartilaginous structures of the ears, nose, upper and lower airways, and ribs, but can also involve joints, skin, eyes, and the cardiovascular system. RP is considered a rare disease (Orpha code: 728) with an estimated incidence of 3.5/1000000/year[1], although a lower figure has been reported in a recent population-based cohort study in the United Kingdom[2]. Peak incidence is in the fifth decade of life (between 40 and 55 years of age), although the disease has also been described in young children and the older people[3]. Its inconspicuous onset can make early diagnosis very difficult, leading to delayed treatment and consequent increased risk of permanent or life-threatening sequelae. Large upper and/or lower airway tract involvement is a common clinical manifestation, occurring in up to 50% of patients over the course of the disease, and is a major cause of morbidity and mortality[4,5]. Moreover, 10% of patients present with tracheobronchial cartilage involvement as the first manifestation[6]. Thickening of the tracheal wall and destruction of the tracheal cartilaginous rings are characteristic in patients with large airway involvement, whereas tracheomalacia is sometimes observed[7]. Here, we report a case of RP complicated with Sjögren's syndrome.
CASE PRESENTATlON
Chief complaints
Cough with expectoration.
History of present illness
The patient was a 47-year-old Chinese female, had developed a cough with expectoration, producing small volumes of mainly white and sticky phlegm, over the 17 mo prior to diagnosis. Outpatient examination showed a 6-min walk distance within normal range. Pulmonary function tests (Figure 1A) showed an FEV1/FVC of 70.73% (84.48% of the predicted FEV1% value of 103.55%), indicating mild obstructive pulmonary ventilation dysfunction. The patient was given routine treatment. Recently, cough and expectoration gradually become worse and she experienced shortness of breath after an event.
History of past illness
The patient denied any history of past disease, allergy, or exposure to smoke or dust for herself or her parents.
Personal and family history
The patient denied any history of past disease, allergy, or exposure to smoke or dust for herself or her parents.
Physical examination
Physical examination only showed rhonchi in both lungs, while other examination parameters were normal.
Laboratory examinations

Figure 1 Respiratory function tests. A: December, 2018; B: May, 2020. Compared with previous measurements, the expiratory flow was impaired and exhibited a platform-like change, suggestive of variable intrathoracic (upper airway) obstruction.
Maximum white and red blood cell counts were 7.58 × 109/L and 4.28 × 1012/L, respectively; neutrophil count was 58%; lymphocyte count was 33%; maximum platelet count was of 349 × 109/L; erythrocyte sedimentation rate was 43 mm/h; patient was anti-Ro52-positive; and mean IgG4 level was 136 mg/L (normal range: 80-1400 mg/L). Other pertinent blood workups (including renal and hepatic parameters, albumin-to-globulin ratio, and levels of C-reactive protein, creatinine kinase, rheumatoid factor, anti-CCP, anti-ENA (including anti-SSA and anti-SSB), MPO, PR3, p-ANCA, and c-ANCA) reported normal values. Arterial blood pH was 7.415; PO2was 87.1 mmHg; PCO2was 32.5 mmHg; and oxygen saturation was 97.3%. Basal salivary flow rate was 0.2 mL/min, reaching 0.5 mL/min after stimulation. The tear film break-up times were 7 s (left) and 10 s (right). Hearing test showed high-frequency hearing loss in the left ear. Upon review of lung function, the forced expiratory volume in 1 s/forced vital capacity (FEV1/FVC) of 38.8% accounted for 47.6% of the expected FEV1% value of 42.6% (Figure 1B). The detailed data of the two lung function tests were compared in Table 1. The airway provocation test was negative, and the bronchial dilation test did not effectively dilate the airway. The Modified Medical Research Council dyspnea scale score was 2.
Imaging examinations
A chest X-ray showed that the texture of both lungs was increased (Figure 2). Chest computed tomography (CT) (Figure 3) revealed thickening of the tracheal wall; bilateral bronchial narrowing in the expiratory phase, compared to the inspiratory phase, was further detected during dynamic breathing. Bronchoscopy (Figure 4) revealed that the tracheal cartilage rings were blurred and the tracheal mucosa was swollen. 18F-fluorodeoxyglucose positron emission tomography/CT (18F-FDG PET/CT) (Figure 5) revealed that the walls of the trachea, the left and right main bronchi, and the lobar and segmental bronchi were evenly thickened and exhibited mild to moderate increases in metabolic activity (SUVmax= 2.91). No abnormal FDG uptake was detected in the paranasal sinuses, nasal cavity, ears, eyes, auricles, or ribs. Biopsy of the lower lip’s salivary gland tissue (Figure 6) exhibited partial ductal dilatation and acinar and periductal infiltration of numerous lymphocytes (> 50 in a focal lesion region).
FlNAL DlAGNOSlS
RP complicated by Sjogren's syndrome.
TREATMENT
Prednisone acetate (40 mg/d).

Table 1 Comparison of main data from respiratory function tests

Figure 2 Chest X-ray. The trachea is in the centre and the textures of both lungs are enhanced.
OUTCOME AND FOLLOW-UP
After treatment with prednisone acetate (40 mg/d), the cough was relieved after 2 wk and the respiratory symptoms disappeared at the 4-mo follow-up.
DlSCUSSlON
RP is diagnosed principally on the basis of its typical clinical manifestations. In 1976, McAdamet al[8] proposed the first diagnostic criteria for RP on the basis of the clinical presentation observed in 159 patients; those criteria were later modified by Damiani and Levine[9] in 1979, and by Michetet al[10] in 1989. In a 2016 report, Dionet al[11] analysed the clinical characteristics of 142 RP patients and defined three clinical phenotypes, a respiratory phenotype, an hematologic phenotype, and a mild phenotype (with good prognosis) that differed in terms of clinical manifestations, disease progression and treatment, and infection rates. Although diagnostic criteria have traditionally focused on rheumatic diseases, since 1972 several RP cases involving valvular heart disease, myasthenia gravis, and myelodysplastic syndrome have been reported, based on histopathological examination after appearance of severe complications[12-16]. Over the last two decades, the medical literature reported also an increasing number of RP cases associated with bronchopulmonary symptoms[17-20]. One prospective study found that among all RP patients, those with predominant respiratory symptoms were younger and had a higher intensive care unit (ICU) admission rate[21]. This evidence suggests the need to expand awareness and understanding of the respiratory manifestations of the disease among clinicians.
RP is a rare disease, characterised by recurring inflammation of cartilage and proteoglycan-rich tissue triggering progressive anatomical deformation and functional damage[1]. As with our patient, about one third of patients with RP may display associated autoimmune diseases[22]. Infiltration of tissues by different cellular and molecular inflammatory mediators leads to the release of degradative enzymes (such as matrix metalloproteinases) and reactive oxygen metabolites by inflammatory cells and chondrocytes, and ultimately to the destruction of cartilage and other proteoglycan-rich structures[23,24]. Autoantibodies against cartilage, collagen (mostly type II, but also types IX, X, and XI), matrilin-1, and cartilage oligomeric matrix proteins have been consistently detected in RP patients[25]. In turn, cytokines released during the inflammatory process can both amplify the pathologic process and induce constitutional symptoms. Based on positive correlation, detected in a single RP patient, between serum Th1 cytokine (e.g., IFN-γ, IL-12, and IL2) levels and disease activity, it was suggested that RP is a Th1-mediated disease[26].

Figure 3 Computed tomography analysis. A and B: The diameters of the trachea and bilateral bronchi show narrowing in the expiratory phase (B) compared to the inspiratory phase (A); C and D: The axial computed tomography image of mediastinal field for inspiration (C) and expiration (D).

Figure 4 Bronchoscopy. The tracheal cartilage ring appears blurred and the tracheal mucosa evidences swelling. A: Tracheal juga; B: Left principal bronchus; and C: Right primary bronchus.

Figure 5 Fluorodeoxyglucose positron emission tomography/computed tomography imaging. A-C: The walls of the trachea and left and right main bronchi show even thickening (with obvious thickening of the tracheal cartilage). Metabolic activities in these structures are slightly to moderately enhanced, especially near the hilum. The metabolic distributions were figure-eight- or strip-shaped. SUVmax = 2.91.
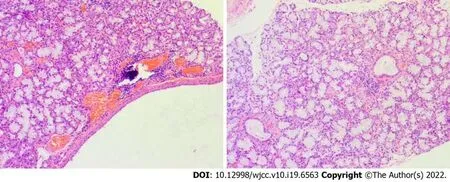
Figure 6 Lower lip’s salivary gland tissue biopsy (magnification, 200 ×). A focal region with > 50 lymphocytes is depicted.
Feared consequences of tracheobronchial compromise in RP patients include structural malformations such as tracheomalacia and permanent tracheal stenosis[6]. Thus, early diagnosis of RP in patients such as ours, presenting with atypical clinical manifestations, is of great importance but also quite challenging. By relying solely on pulmonary function tests indicating obstructive airflow limitations, a diagnosis of RP can be easily confused with several other conditions in patients presenting only with respiratory symptoms such as cough and dyspnea. In turn, the presence of respiratory symptoms in a patient already diagnosed with RP should raise clinical suspicion of potentially severe airway lesion and prompt further testing and treatment.
Given the non-specific, airway-delimited pathological manifestations of our patient, a diagnosis of RP was initially missed upon applying the aforementioned testing criteria. However, the sharp decline in pulmonary function experienced by our patient (progressing from mild to severe within 18 mo) served as a red flag, and we thus made every effort to perform a correct diagnosis. Expiratory CT abnormalities are present in the majority of RP patients, yet only half of them demonstrate abnormalities on routine inspiratory CT scans[27,28]. In our clinical cases, chest CT objectively reflects airway wall thickening, but does not clearly define the extent of stenosis or obstruction. Pulmonary function tests and chest CT are thus complementary, but the former are more informative. Inflammation of the mucosa and infiltration of the cartilaginous structures are sometimes visible on endoscopy above the thyroid and next to the first tracheal rings. We used FDG-PET/CT to evaluate cartilage metabolism at other sites (as an alternative to biopsy) since this technique proved to be a useful tool for both diagnosis and evaluation of disease activity and has been used for diagnosis of RP[29,30].
In our patient, we detected no abnormal FDG uptake in the nose, ears, or other cartilage-rich areas. Instead, we found that metabolic activities in the trachea and left and right main bronchi were slightly to moderately enhanced, and the cartilage of the tracheal wall was evenly thickened. Tracheobronchial amyloidosis was hence ruled out, because it typically involves inhomogeneous nodular thickening. Tracheal cartilage biopsy is sometimes indicated in patients with RP to visualize lesions and structural anomalies undetected by CT; however, invasive bronchoscopy may aggravate mucosal swelling or cartilage inflammation, triggering airway spasm and even severe or fatal respiratory distress[6]. Considering the patient’s radiographic findings and the marked deterioration in lung function occurring over the preceding 18 mo, a diagnostic biopsy was ruled out. Aided by improved diagnosis and treatment, the prognosis of RP patients has steadily improved, with 10-year survival rates of 55% reported in the 1980’s and 8-year and 10-year survival rates of 94% and 91% reported in 1998 and 2016, respectively[6].
Although patients with severe tracheobronchomalacia can be successfully treated with bronchoscopyguided intervention therapy[31], we have doubts about the efficacy of the treatment in those patients who have, like ours, pathological laryngopharyngeal reflux. Our patient is currently stable, reports a high quality of life, and is under long-term follow-up to monitor treatment efficacy.
CONCLUSlON
In conclusion, we present a case-report on a patient with RP with airway involvement as the only clinical manifestation and provide an account of the difficulties encountered in establishing the correct diagnosis. We learned from this case that PET/CT and bronchoscopy can help confirm a diagnosis of RP when common disease signs and symptoms are not obvious or absent. Currently, there are no standardized guidelines for RP diagnosis, which is mostly based on clinical manifestations and symptom-driven diagnostic testing[32]. Because RP patients with airway involvement may have higher infection risk and more commonly require admission to an ICU, it is pivotal to recognize and manage the airway morbidities in a timely manner to prevent fatal consequences.
ACKNOWLEDGEMENTS
We are thankful to the patient for her kind permission to report the clinical presentations and the laboratory and radiographic data related to her illness.
FOOTNOTES
Author contributions:Chen JY and Li XY wrote the first draft of the manuscript; Zong C wrote additional sections of the manuscript; all authors contributed to manuscript revision, read, and approved the submitted version.
lnformed consent statement:The patient's consent and informed consent were obtained for this case report.
Conflict-of-interest statement:The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
CARE Checklist (2016) statement:The authors have read the CARE Checklist (2016), and the manuscript was prepared and revised according to the CARE Checklist (2016).
Open-Access:This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BYNC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is noncommercial. See: https://creativecommons.org/Licenses/by-nc/4.0/
Country/Territory of origin:China
ORClD number:Jun-Yan Chen 0000-0002-3347-6799; Xiao-Yan Li 0000-0001-7268-9853; Chen Zong 0000-0002-5263-0253.
S-Editor:Gong ZM
L-Editor:A
P-Editor:Gong ZM
 World Journal of Clinical Cases2022年19期
World Journal of Clinical Cases2022年19期
- World Journal of Clinical Cases的其它文章
- Current guidelines for Helicobacter pylori treatment in East Asia 2022: Differences among China, Japan, and South Korea
- Review of epidermal growth factor receptor-tyrosine kinase inhibitors administration to non-small-cell lung cancer patients undergoing hemodialysis
- Arteriovenous thrombotic events in a patient with advanced lung cancer following bevacizumab plus chemotherapy: A case report
- Endoscopic ultrasound radiofrequency ablation of pancreatic insulinoma in elderly patients: Three case reports
- Acute choroidal involvement in lupus nephritis: A case report and review of literature
- Choroidal thickening with serous retinal detachment in BRAF/MEK inhibitor-induced uveitis: A case report