
数据挖掘探究肝-肾-肺纤维化的关键基因
2022-12-17 03:21:26闫慧芳
生物信息学 2022年4期
闫慧芳
(中国人民大学 统计学院,北京 100872)
器官纤维化是细胞外基质(Extracellular Matrix, ECM)的过度积累,从而导致组织结构的扭曲及器官功能的丧失。很多慢性病可引起纤维化,包括糖尿病、高血压、病毒性、慢性肝病等疾病导致的纤维化可因过量的ECM取代和破坏实质组织而致肝、肺、肾、心脏或其他重要器官的衰竭[1]。最近的分析表明,在西方发达国家,接近一半的死亡都与纤维性疾病有关[2]。解决器官纤维化问题的能力可能因为一些因素而不同,包括器官、有害刺激因素的性质以及患者的特征(例如年龄和遗传背景)[3-5]。然而,来自人类和动物模型的很多研究表明,纤维化在所研究的大多数器官或组织中存在被逆转的可能。
计算生物学方法在确定人类疾病的潜在遗传基础方面起着至关重要的作用[6]。高通量平台例如微阵列技术和RNA测序技术可用于分析转录层面上基因的改变,越来越被视为在疾病研究中提供重要帮助的工具[7-9]。近年来,结合生物信息学分析的微阵列技术能够快速处理大量的数据集,使人们能够全面识别数百个与某些疾病的发生和发展有关的差异表达基因(Differential Expressed Genes,DEGs)。
针对器官纤维化也有许多研究工作,它们从不同的角度阐释了不同组织间纤维化的共同特征。虽然组织成纤维细胞通常是异质性的,但病理性肌成纤维细胞在肺、肾或肝纤维化中表现出类似的组织学表型和分子特征,这表明基本的致病途径在不同纤维化器官存在共性[10-12]。Zhu Z.等整合了3个微阵列公共数据集,利用生物信息分析识别了肝纤维化相关的基因和通路,此外还对潜在的治疗药物进行了初步筛选[13]。Chau-Chyun S.等针对特发性肺纤维化利用二代测序(Next Generation Sequencing, NGS)平台获取纤维化肺和健康肺的mRNA数据,利用逐步的生物信息学手段分析获得与之相关的差异基因,他们也利用了Gene Expression Omnibus (GEO)公共数据集进行了验证[14]。Wang S.等识别出CXCL14 是一个由不同疾病引发的肝纤维化的共有差异基因,它可能会成为治疗多因素肝纤维化的潜在靶点[15]。Mira P.等利用实验收集了肾纤维化相关的多组学数据(包括DNA,miRNA和蛋白),利用计算生物学方法进行了整合分析,并将这些数据和分析结果生成了在线工具,以供研究人员查找肾纤维化相关的转录组和蛋白组信息[16]。Zhao J.Q.等对纤维化的心血管疾病进行了差异基因分析和功能富集,最终识别出11个关键基因[17]。Eugene M.等利用计算方法分析了肝纤维化和肺纤维化的共有代谢通路,为探究纤维化治疗方法提供了新思路[18]。虽然近年来研究人员已经利用生物信息学对纤维化进行了一系列研究,仍有以下几方面需要进一步探究:1)不同器官纤维化的共有关键基因尚不完全明确;2)肝纤维化的特有关键基因信息鲜有研究;3)此外,为了发现潜在药物靶点,针对关键基因及蛋白的成药性分析是十分必要的[19]。
本文利用计算生物学方法对GEO公共数据集 (GSE36066, GSE97546, GSE55747) 进行了整合分析和数据挖掘。其中GSE36066和 GSE97546包含 肝、肾、肺各一组数据,先基于它们进行了第一轮分析。之后另外引入一个肝纤维化数据集GSE55747做进一步的验证。最后针对验证后的纤维化关键基因,进行了成药性的评估。这些研究将有助于进一步理解纤维化过程的内在机理,也可为纤维化生物标志物和潜在药物靶点的发现提供借鉴。
1 材料与方法
1.1 数据集
从GEO数据库中下载3个基于小鼠的mRNA数据集,其中GSE36066和GSE97546用于差异表达基因和相关功能富集等的分析,GSE55747用于结果的验证。3个数据集的基本信息(见表1)。其中GSE36066为胆管结扎诱发的肝纤维化样本与非纤维化样本,GSE97546包含单侧输尿管梗阻诱发的肾纤维化和博来霉素诱发的肺纤维化样本及相应的对照样本,验证数据集GSE55747则是四氯化碳诱发的肝纤维化样本与非纤维化肝样本。3个数据集的采集基于不同的微阵列平台,数据样本的对照组和处理组样本均有生物学重复。

表1 数据集的基本信息Table 1 Basic information of datasets
1.2 差异表达基因(Differentially Expressed Genes, DEG)的分析及选择
GEO2R(http://www.ncbi.nlm.nih.gov/geo/geo2r)提供了友好用户界面,能够对GEO数据进行基于R的统计分析。 GEO2R是一种智能的在线分析工具,可以在相同的实验条件下既能整合分析两个数据集,也可以拆分任何GEO数据[20]。本研究应用GEO2R工具分析纤维化器官与正常器官之间差异表达基因。调整p值 < 0.01,log2FC(Fold Change)的绝对值 > 1.0的基因被认为是差异表达基因DEGs。获取差异表达基因后利用在线Venn分析工具(http://bioinformatics.psb.ugent.be/webtools/Venn)获得肝、肾、肺纤维化的DEG交集及肝纤维化特有DEG。
1.3 GO(Gene Ontology)功能富集及KEGG信号通路分析
基因本体论(Gene Ontology, GO)是一种常用的生物信息学工具,可根据定义的特征提供有关单个基因组产品的基因功能的全面信息。GO富集分析能从三方面更好地解释基因的功能:分子功能(Molecular Function, MF),生物过程(Biological Process, BP)和细胞成分(Cellular Component, CC)。KEGG(Kyoto Encyclopedia of Genes and Genomes, KEGG)是系统分析基因产物在细胞中的代谢途径以及这些基因产物功能的数据库。本研究中,使用Webgestalt(http://www.webgestalt.org)进行了GO和KEGG分析[21]。
1.4 验证分析
利用数据集GSE55747对以上分析获取的差异基因进行验证。首先使用GEO2R在线平台分析获得该数据集中肝纤维化样本与对照样本的差异表达基因。调整p值<0.01,log2FC(Fold Change)的绝对值>1.0的基因被认为是差异表达基因DEGs。然后利用Venn分析工具对GSE55747的DEGs与先前识别出的三种器官纤维化共有差异基因和肝纤维化特有差异基因进行比较分析,取两者的交集,从而得到进一步验证的差异表达基因列表。
1.5 蛋白质-蛋白质相互作用 (Protein-Protein Interaction, PPI)及关键基因识别
STRING(Search Tool for the Retrieval of Interacting Genes , STRING)[22]数据库提供了实验和预测的相互作用信息。在本研究中,STRING在线工具用于分析DEG综合得分 > 0.4的PPI。然后通过Cytoscape可视化PPI网络[23],利用其中的CytoHubba插件[24]分别计算得到3种器官纤维化共差异基因和肝纤维化特有差异基因PPI网络中前10个具有最高连接度的基因(蛋白),将其视为关键基因(蛋白)。
1.6 关键基因/蛋白的成药性(Druggability)评估
本研究主要基于3个维度的信息评估关键基因/蛋白的成药性。以往的研究表明药物-蛋白相互作用的发生区域对于药物的合理递送是十分重要的[25],因此我们将基因/蛋白的分布区域作为评估的第1个维度,如果蛋白分布于胞外或细胞膜,则赋分2分,其他分布区域赋分为0分。通常分泌蛋白的性质会利于该蛋白的成药性[26]。因此是否有证据表明蛋白为分泌蛋白,是评估的第2个维度,分泌蛋白赋分1分,非分泌蛋白赋分0分。此外有些蛋白并没有直接的证据表明它们分布于胞外或为分泌蛋白,但是曾有研究在血浆中检测到它们的存在,这一信息也是成药性的因素之一。本研究将血浆中曾检测到浓度的蛋白赋分1分,其余赋分0分。最后对各关键蛋白/基因的三项赋分加和得到最终的成药性评分,0-4分代表成药性由低到高。所获取的蛋白分布、是否为分泌蛋白等信息均来自于HPA(The Human Protein Atlas) 数据库[27]。
2 结果分析
2.1 多器官纤维化共有差异表达基因(DEGs)及肝纤维化特有差异表达基因
利用GSE36066和GSE97546数据集,识别了肝、肾、肺三种器官纤维化的各自的差异表达基因。adj.p值 < 0.01,log2FC(Fold Change) > 1.0作为界定差异基因的标准。通过GEO2R分析,GSE36066肝纤维化DEG 2 382个,GSE97546肾纤维化 DEG 2 632个,GSE97546肺纤维化DEG 2 897个 (见图1)。随后对三种器官纤维化的DEG进行韦恩分析,得到多器官纤维化的共有DEGs 196个和肝纤维化特有DEGs 1 562个(见图2)。

图1 差异表达基因的火山图Fig. 1 Volcano diagram of DEGs
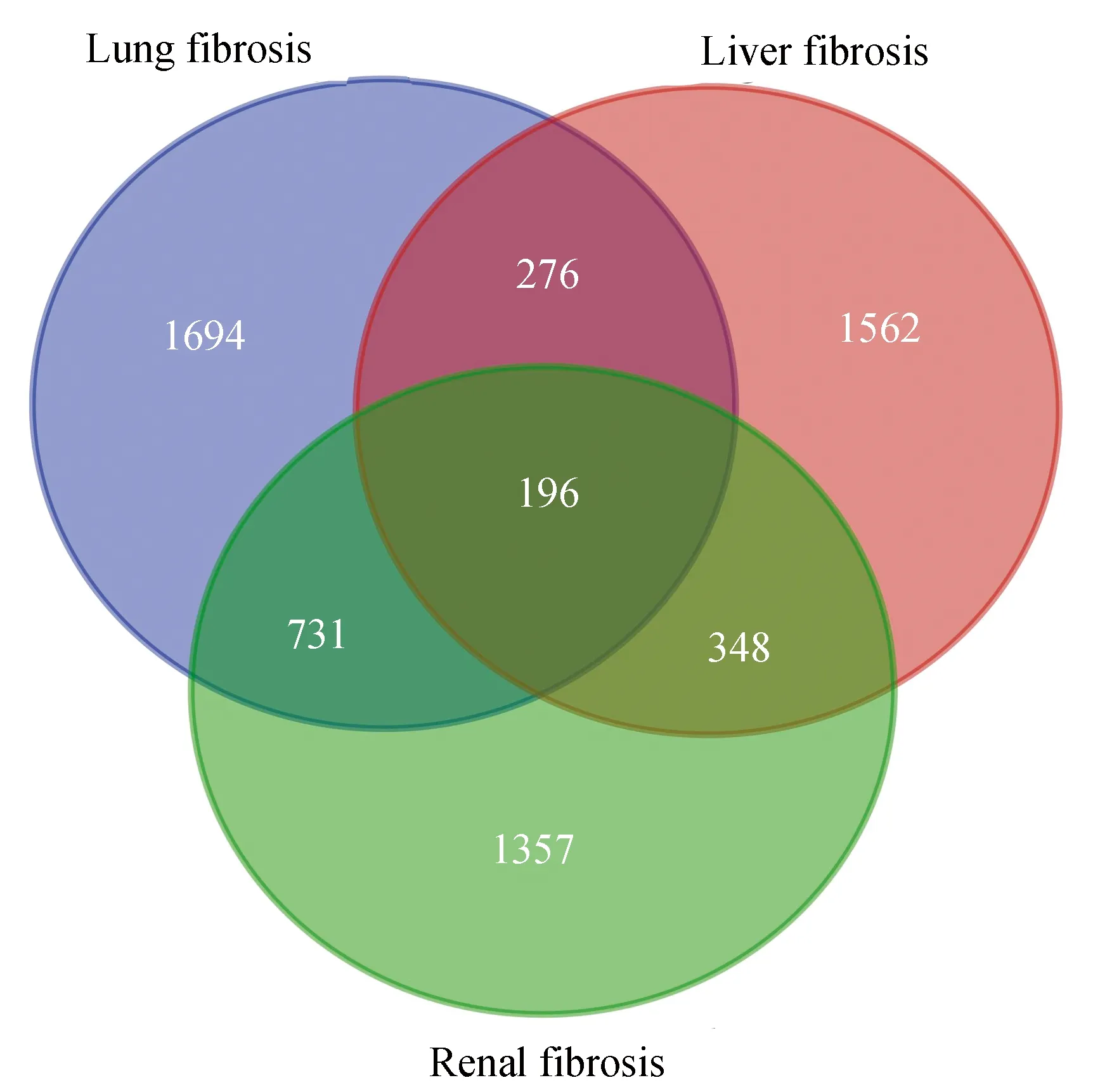
图2 差异表达基因的韦恩图Fig. 2 Venn diagram of DEGs
2.2 GO功能富集和KEGG通路
为了进一步理解差异表达基因参与的生物过程和信号通路,利用Webgestalt在线工具对多器官纤维化共有DEG进行了GO功能富集和KEGG信号通路分析,另外对肝纤维化DEG进行了生物过程的功能富集分析。
2.2.1 三种器官纤维化共有差异基因的功能富集和信号通路分析
对肝、肺、肾三种器官纤维化共有的196个差异基因进行的GO功能富集和KEGG信号通路分析结果(见图3)。采用有向无环图(DAG)展示GO功能富集的分析结果,其中生物过程(BP)富集最显著的包括炎症反应、免疫反应、免疫系统调节等(见图3a)。分子功能(MF)富集显著的为脂磷酸结合、趋化因子活性、信号受体结合等(见图3c)。而细胞组成(CC)分析显示,这些基因表达的蛋白大部分为细胞外区域、细胞表面或细胞外壁的组分,这也一定程度上为蛋白/基因的成药性提供了证据(见图3b)。此外,KEGG通路分析的火山图(见图3d) 显示差异表达基因在TNF信号,Toll受体信号、流体剪切应力与动脉粥样硬化等通路中有较显著的富集。

图3 GO功能富集和KEGG信号通路分析Fig. 3 GO functional enrichment and KEGG signaling pathway analyses
2.2.2 肝纤维化特有差异基因的生物过程(BP)功能富集
对肝纤维化特有的1 562个差异基因进行的生物过程(BP)富集分析结果(见图4)。BP富集分析中满足p值和错误发现率(False Discovery Rate, FDR)均 < 0.05的生物过程被视为显著,然后按照富集比率(Enrichment Ratio, ER)由大到小排列取前10项。结果表明这些差异基因的生物功能主要集中在脂肪酸代谢、羧酸代谢、脂质代谢、氧化还原等过程。

图4 肝纤维化特有差异基因的生物过程富集Fig.4 Biological process enrichment for liver fibrosis specific DEGs
2.3 数据验证获得最终的差异表达基因
本研究除了探究三种器官纤维化共有关键基因以外,还研究了肝纤维化的特有关键基因,故引入一个肝纤维化数据集GSE55747做进一步的验证。利用GEO2R对数据集GSE55747进行差异基因分析,同时满足adj.pvalue<0.01 和log2FC>1.0被认为是显著的差异表达基因DEG。然后利用Venn分析工具与先前得到的肝肾肺三器官纤维化共有差异基因(196个)和肝纤维化特有差异基因(1 562个)分别取交集。最终得到验证后的肝-肾-肺三器官纤维化共有差异基因58个,肝纤维化特有差异基因85个(见表2)。

表2 验证后的三器官纤维化共有差异基因和肝纤维化特有差异基因Table 2 Common DEGs of fibrosis in three organs and liver fibrosis specific DEGs after validation
2.4 蛋白质-蛋白质相互作用(PPI)及关键基因
为了更好地理解识别得到的共有差异基因和特有差异基因中哪些起到关键作用,利用STRING工具进行了蛋白质-蛋白质相互作用的分析,得到了包含58个nodes、145个edges的共有差异基因网络和包含82个nodes、53个edges的肝纤维化特有差异基因网络(见图5)。然后利用Cotoscape中Cytohubba计算插件分别对这2个PPI网络得到基于MCC (Maximal Clique Centrality) 法排序前10的基因。本研究中将这些基因作为与纤维化过程相关的肝肾肺共有关键基因和肝特有关键基因(见表3)。将这20个小鼠基因与人类基因进行匹配,得到14个匹配成功的对应人类基因。
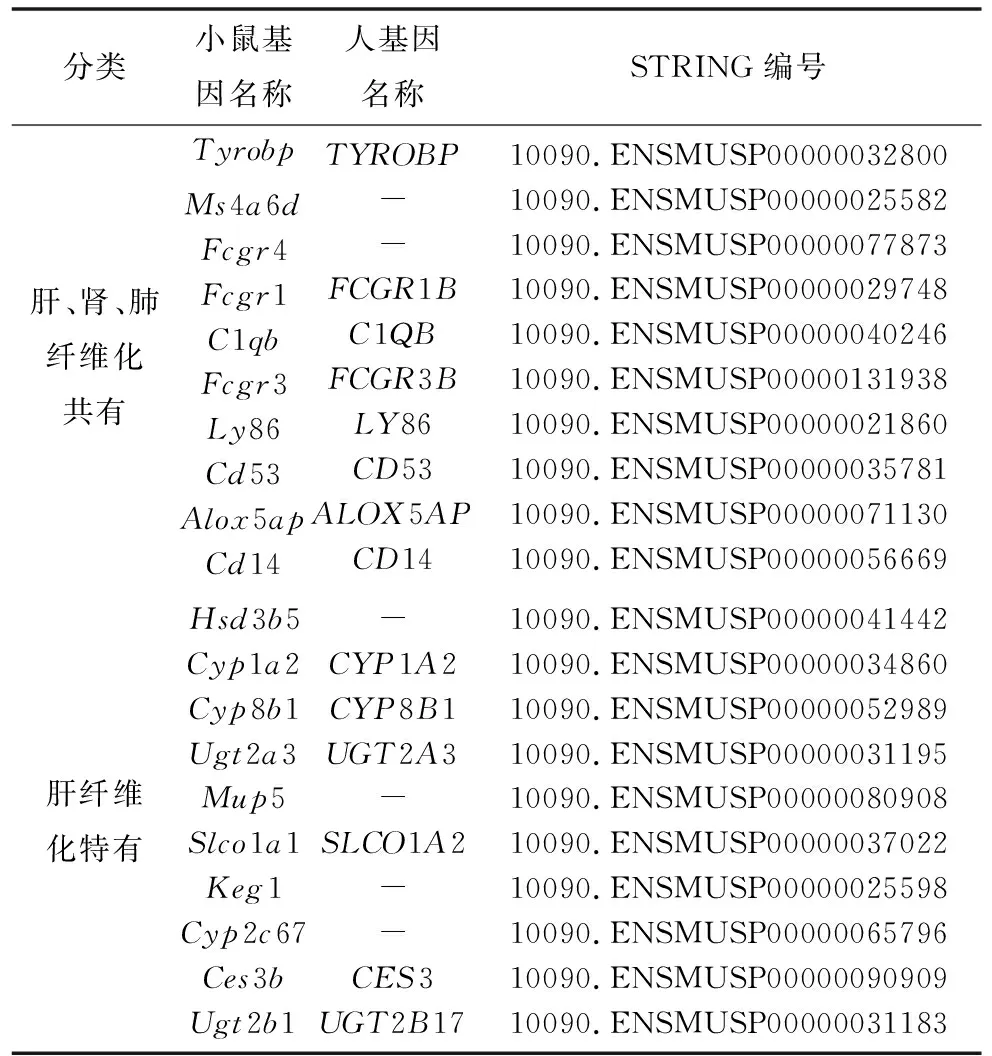
表3 多器官纤维化和肝纤维化特有的关键基因 (各取前10个)Table 3 Hub genes for multi-organ fibrosis and liver fibrosis only (ten for each)

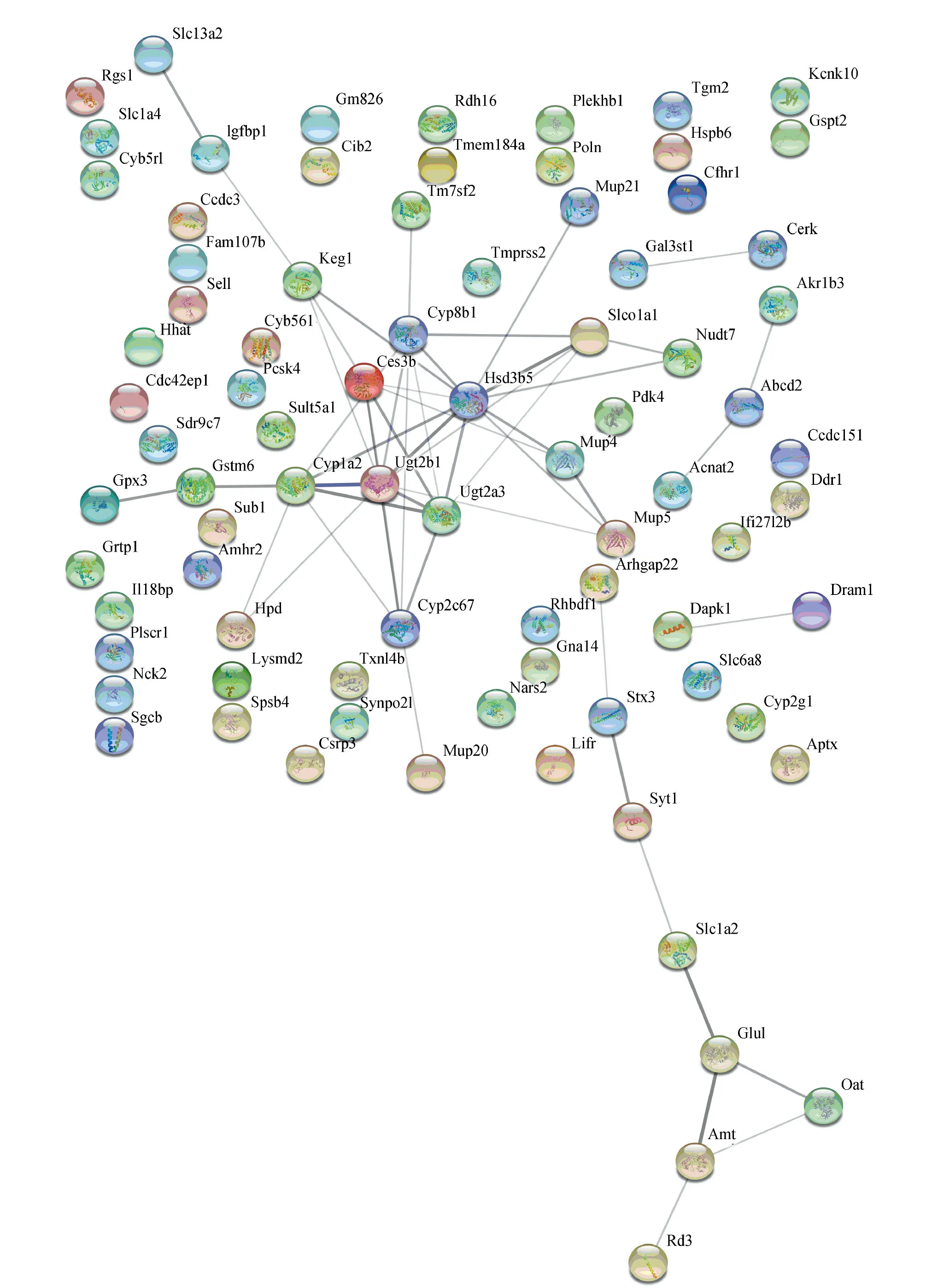
图5 蛋白质-蛋白质相互作用网络Fig.5 Protein-protein interaction network
2.5 关键基因的成药性
蛋白分布区域、是否为分泌蛋白以及是否有研究在血浆中检测到为3个考虑的关键指标,这些指标对应的信息全部来自于公共数据库。针对这些信息,进行相应的赋分,其中分布区域为成药性最重要的因素,因此分布于胞外或细胞膜则赋2分。如果为分泌蛋白或有血浆中浓度信息各赋1分。将这三项的赋分加和,即得到最终的成药性评分。经过分析计算,在这14个关键基因中,有3个具有较高的成药性(4分),6个有中等的成药性(2~3分), 5个具有较弱的成药性(0~1分),具体结果(见表4)。
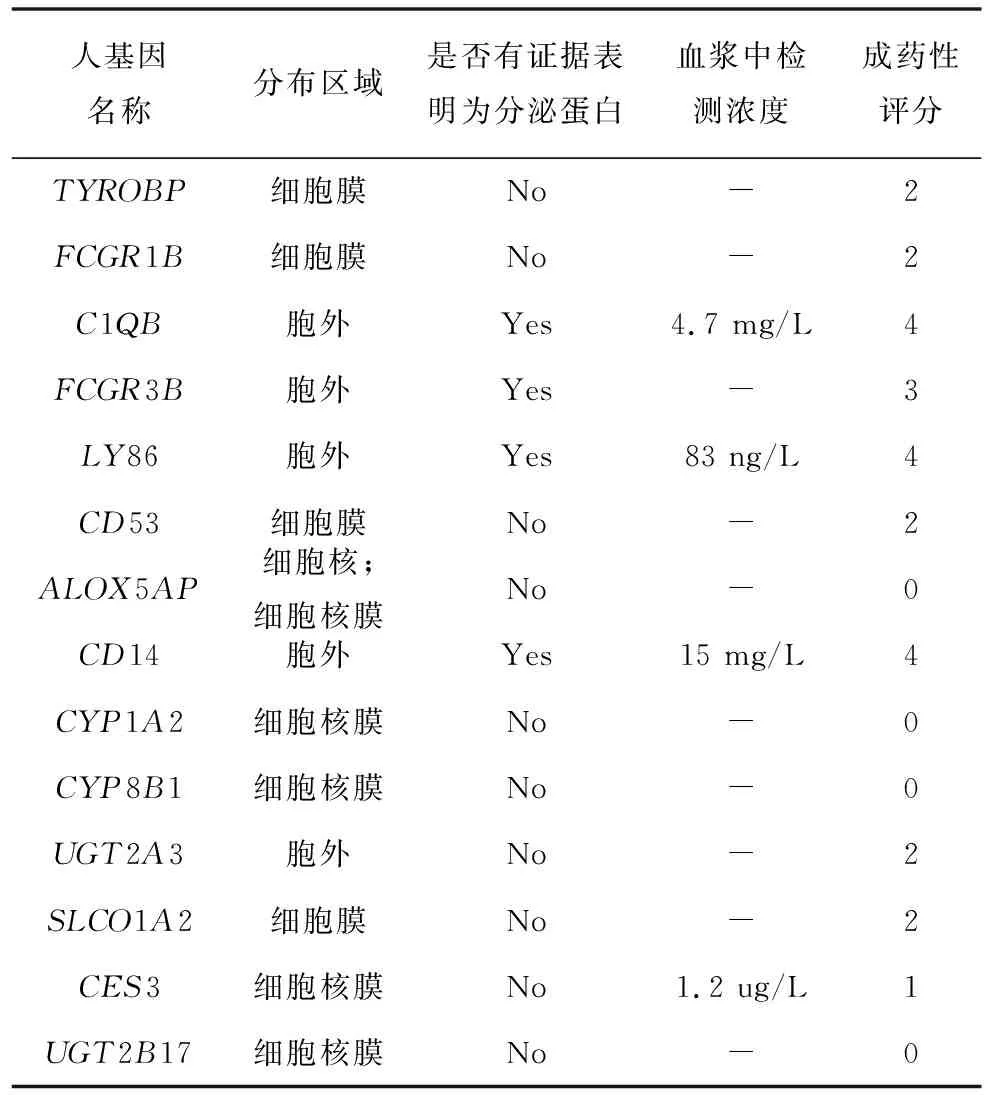
表4 关键基因的成药性信息Table 4 Druggability information for hub genes
3 讨 论
纤维化是细胞外基质的过度积聚,通常是由于重复或慢性组织损伤的伤口愈合反应,并可能导致器官结构破坏和功能的丧失。虽然纤维化在以前被认为不可逆,最新的证据表明某些情况下纤维化疾病是可以被逆转的,纤维化消退的机制包括降解纤维化的细胞外基质以及消除相关的成纤维细胞。已有研究表明不同器官的纤维化机制存在共性,因此为了探究与纤维化过程相关的关键基因和信号通路,本研究通过对公共数据集的数据挖掘,识别了肝脏、肾脏及肺脏三种器官纤维化的共有关键基因和相关生物通路。此外,也对肝纤维化的特有差异基因进行了分析。研究人员做了大量基于动物模型的纤维化研究,虽然动物模型并不能完全描述相应的人类疾病,但是相比于人的试验,动物试验具有更加可控可比的样本组和对照组,也利于采集噪音更小的数据。已经有许多具有代表性的纤维化动物模型。其中胆总管结扎和四氯化碳诱导作为代表性的肝纤维化动物模型构建方法。胆管结扎动物模型在全世界数百个实验室中用于诱发肝胆汁淤积和纤维化。它诱导肝内胆管上皮细胞增生,使增殖的胆管上皮细胞周围的门脉成纤维细胞发生肌纤维母细胞分化,从而导致ECM的高度复制,大量表达和沉积。因此,该模型在大鼠和小鼠中的应用在研究肝炎症和纤维化发病机理的十分流行。四氯化碳诱导的动物模型主要机制为四氯化碳在肝脏中被细胞色素P450代谢,转化为高反应性三氯甲基(CCl3)自由基,最终导致肝毒性损害、炎症和纤维化。CCl4诱导的肝损伤构成了人类病理学的可靠模型,在生理和细胞水平上都模仿了其关键特征。对于肾纤维化,单侧输尿管梗阻(UUO)会在阻塞的肾脏中引发一系列事件,导致肾血流量和肾小球滤过率在24 h内急速下降。随后的间质反应包括间质炎症,肾小管扩张和肾小管凋亡,并从7 d开始导致肾小管萎缩和纤维化。尽管完全的输尿管梗阻不是人类肾脏疾病的最常见原因,但由于UUO具有产生进行性肾纤维化的能力,它已成为慢性肾脏疾病的最受欢迎模型之一。完整的梗阻模型的优点是可重复性好(因此动物间的差异不成问题),时程短,性能容易,并且有对侧肾脏作为对照。此外,该模型在大鼠和小鼠中都很容易诱导。而博来霉素诱导的动物模型是最具代表性的肺纤维化实验模型。博来霉素是一种治疗用的抗生素。当淋巴瘤患者静脉注射BLM后出现肺纤维化时,已被确定为促纤维化药物。它已被用于多种物种的模型构建,包括小鼠,大鼠,豚鼠,仓鼠,狗和灵长类动物。本研究中选用基于以上动物模型收集的数据集,利用GSE36066(肝纤维化)和GSE97546(肾纤维化和肺纤维化)识别出肝-肾-肺纤维化共有差异表达基因196个,肝纤维化特有差异表达基因1 562个。然后引入数据集GSE55747进行了验证分析,得到验证后的共有DEG 58个,肝纤维化特有DEG 85个。肝-肾-肺纤维化共有DEG主要富集于炎症反应(Inflammatory Response)和免疫反应(Immune Response)过程。以往的很多研究表明,化学物质、微生物或生理组织损伤引起的慢性炎症通常会导致纤维化病变[28-30],本研究的富集结果与这一结论相吻合。尚未有研究表明存在未发生炎症反应而产生的纤维化,而不同原因和不同器官中发生的纤维化通路可能十分相似[31]。生物过程(BP)富集结果显示196 个肝肾肺共有DEG中约50个被归类于免疫反应或炎症反应过程。其中CCL5、CCL6、CCL12、CXCL1、CXCL14和CX3CL1属于趋化因子(Chemokines)超家族,趋化因子是一类参与免疫调节和炎症过程的蛋白[32]。研究表明趋化因子参与所有的伤口愈合反应且在纤维化过程中起到通用的及器官特异性的作用[33]。CD14、CD44和CD84基因编码的蛋白都属于表面抗体,优先在巨噬细胞和白细胞上表达。CD14与其他蛋白质协同介导免疫应答[34]。CD44编码的蛋白质是一种细胞表面糖蛋白,参与细胞间相互作用,细胞粘附和迁移。已被证实CD44与其配体HA与许多炎症性疾病有关[35]。CD84编码一种膜糖蛋白,该蛋白是信号淋巴细胞激活分子(SLAM)家族的成员。研究显示CD84介导的信号调节多种免疫过程[36]。C1qa和C1qb分别编码血清补体亚成分C1q的A链和B链多肽,而C1q与自身免疫和细胞凋亡密切相关[37]。
肝纤维化特有DEG的富集分析显示与脂肪酸代谢(Fatty acid metabolic process)和脂质代谢(Lipid metabolic process)过程有很强的联系。富集分析表明1 562 个肝纤维化特有DEG中约160个被归类于脂质代谢或脂肪酸代谢过程。肝脏为脂质代谢的重要器官,而数据集GSE36066和GSE55747中肝纤维化小鼠模型分别基于胆管结扎(BDL)和四氯化碳(CCL4)诱导。以往的研究表明,BDL诱导会降低肝脏中细胞色素P450含量、过氧化脂肪酸的beta-氧化和催化活性,而这些变化并不会发生在肾脏中[38]。也有很多研究表明CCl4会引发脂质过氧化,从而造成肝损伤[39-42]。因此相较于肾脏和肺脏纤维化,在肝纤维化中特异性地识别出脂质代谢相关的差异基因是合理的。而这些与脂质代谢或脂肪酸代谢相关的基因是否特异性地在肝纤维化中发挥作用,还需要进一步的验证与分析。
利用PPI分析得到肝-肾-肺纤维化共有和肝纤维化特有关键基因各10个,经过小鼠基因与人基因的匹配,成功识别出肝-肾-肺纤维化共有的人关键基因8个,肝纤维化特有的人关键基因6个。通过文献调研发现,此前的研究有证据表明这些关键基因中5个与纤维化相关(TYROBP,FCGR3B,ALOX5AP,CD14,CYP1A2),3个与肝病相关(CYP8B1,UGT2A3,CES3)。TYROBP(DAP12)编码包含基于免疫受体酪氨酸激活模体的跨膜信号多肽,参与淋巴与非淋巴细胞免疫调节的相互作用。Zhang L.等的研究表明在原发性胆汁性肝硬化(PBC)中TYROBP基因下调[43]。Tammaro A.等则发现TYROBP(DAP12) 参与了UUO诱发的肾纤维化[44]。FCGR3B编码的蛋白是针对γ免疫球蛋白(IgG)Fc区的低亲和力受体。它能够充当单体,与单体或聚集的IgG相结合,可能在捕获外周循环中免疫复合物过程中起到一定作用。Bournazos S等在一项病例对照研究中,使用CD36作为基因复制对照,通过实时定量PCR比较了142例IPF患者和221例对照中FCGR3B的拷贝数。证明了该基因拷贝数的差异(Copy Number Variation, CNV)与特发性肺纤维化有关[45]。ALOX5AP编码一种是白三烯合成所必需的蛋白,白三烯是花生四烯酸代谢产物,已经证实与哮喘、关节炎等多种炎症反应有关。针对囊性纤维化疾病,靶向ALOX5AP的药物Fiboflapon在临床二期研究中。另外Kowal-Bielecka O等的研究表明ALOX5AP的遗传变异可能在肺纤维化中起作用[46]。CD14编码的蛋白属于表面抗体,优先在巨噬细胞和白细胞上表达,CD14与其他蛋白质协同介导免疫应答反应。Zhao SX等人的研究表明CD14阳性的单核细胞与 CD163阳性的巨噬细胞慢性丙型肝炎患者肝纤维化的严重程度相关[47]。Fukushima H的研究则表明,在具有纤维化非酒精性脂肪肝炎(NASH)的动物模型中,CD14阳性的肝巨噬细胞数量增加[48]。CYP1A2编码一种细胞色素P450酶, 细胞色素P450是单加氧酶,可催化涉及药物代谢和胆固醇和其他脂质合成的许多反应。Wuensch T等在非肿瘤肝组织中,观察到CYP1A2活性逐渐下降,并伴有纤维化加剧[49]。在肝特异相关的关键蛋白中,CYP8B1在此前的研究中被证明与非酒精性脂肪肝病NAFLD(Non-alcoholic Fatty Liver Disease)相关。 Raphael C等通过研究发现在胆固醇诱导的NAFLD模型中,敲低Cyp8b显著降低了脂肪变性和肝脂质含量[50]。Hardwick RN 等发现UGT2A3在非脂肪非硬化的非酒精性脂肪肝炎NASH病人中,表达水平增高,而在脂肪性NASH和肝硬化病人中并未发现这种差异[51]。Quiroga AD在大鼠早期肝癌中发现了CES3基因的下调,证明该基因可能与肝癌的发生发展有关[52]。
研究疾病的机制和关键基因的主要目的是开发更有效的药物或治疗手段。而这些关键基因作为潜在药物靶点的一个重要前提是较高的成药性 (Druggability)。基于蛋白分布区域、是否为分泌蛋白以及检测到的血浆浓度进行的成药性的分析显示,7个共有关键蛋白/基因和2个特有关键蛋白/基因具有较高的成药性。综合富集结果、蛋白-蛋白相互作用、研究证据和成药性信息,结果显示TYROBP, FCGR3B, ALOX5AP和CD14或可作为纤维化治疗的潜在靶点, 而CYP8B1和UGT2A3可能成为NAFLD或NASH等肝病治疗的研究重点。对于本研究中识别出的关键基因FCGR1B, C1QB, LY86和CD53,虽然此前未有研究表明与纤维化直接相关,也可作为全新的靶点进行更深入的探究和验证。
综上所述,本研究利用计算生物学方法对GEO公共数据集进行了整合分析和数据挖掘,识别了肝脏、肾脏及肺脏三种器官纤维化的共有关键基因和肝纤维化的特有关键基因。GO功能富集和KEGG通路分析表明,肝-肾-肺纤维化共有DEG主要富集于炎症反应和免疫反应过程,而肝纤维化特有DEG脂肪酸代谢和脂质代谢过程有很强的联系。最后还对这些关键基因进行了成药性的评估。综合富集结果、蛋白-蛋白相互作用、研究证据和成药性信息给出了纤维化和肝病的潜在药物靶点。这些研究将有助于理解纤维化过程的内在机理,也为纤维化疾病的治疗提供更多的可能。本研究全部基于动物模型公共数据集的分析和挖掘,并未经过实验结果的验证,未来通过体外和体内试验对这些基因进行更深入的研究将有助于进一步明确它们在纤维化过程中发挥的作用。
4 结 论
1)TYROBP,FCGR3B,ALOX5AP和CD14或可成为纤维化治疗的潜在靶点;
2)CYP8B1和UGT2A3可能与非酒精性脂肪性肝病(Non-alcoholic Fatty Liver Disease, NAFLD)或非酒精性脂肪性肝炎(Non-Alcoholic SteatoHepatitis , NASH)相关;
3)对于基因FCGR1B,C1QB,LY86和CD53,目前没有直接的证据表明其与纤维化相关,需要进一步验证它们在纤维化发生过程中的功能。
猜你喜欢
动物医学进展(2024年4期)2024-04-10 01:50:04
中老年保健(2022年2期)2022-11-25 23:46:31
昆明医科大学学报(2022年4期)2022-05-23 13:04:50
基层中医药(2020年12期)2020-07-22 06:35:00
心电与循环(2020年1期)2020-02-27 07:48:24
国际呼吸杂志(2019年21期)2019-11-25 09:52:20
中国资源综合利用(2016年10期)2016-01-22 08:36:17
中国卫生标准管理(2015年25期)2016-01-14 09:29:27
天津医科大学学报(2015年3期)2015-06-05 12:21:49
电测与仪表(2015年10期)2015-04-09 11:48:04
